BARACLUDE
B-MS
entecavir
Antiviral.
Apresentações.
BARACLUDE comprimidos revestidos é apresentado nas concentrações de 0,5 mg e 1 mg, em frascos contendo 30 comprimidos.
USO ORAL
USO ADULTO E ADOLESCENTE ACIMA DE 16 ANOS
Composição.
Cada comprimido revestido contém 0,5 mg ou 1 mg de entecavir, respectivamente. BARACLUDE comprimidos revestidos contém os seguintes ingredientes inativos: lactose monoidratada, celulose microcristalina, crospovidona, povidona e estearato de magnésio. O revestimento do comprimido contém dióxido de titânio, hipromelose, polietilenoglicol 400, polissorbato 80 (apenas comprimido de 0,5 mg) e óxido de ferro vermelho (apenas comprimido de 1 mg).
Indicações.
BARACLUDE é indicado para o tratamento de infecção crônica pelo vírus da hepatite B1 (VHB) em adultos com evidência de replicação viral ativa e também com evidências de elevações persistentes nas aminotransferases séricas ALT e AST ou doença histologicamente ativa.
1CID B18.1 - Hepatite crônica viral B sem agente Delta
Os seguintes pontos devem ser considerados quando é iniciada a terapia com BARACLUDE:
- Esta indicação se baseia nas respostas histológicas, virológicas, bioquímicas e sorológicas em pacientes adultos virgens de tratamento com nucleosídeos e pacientes adultos resistentes à lamivudina com infecção VHB crônica com HBeAg-positivo ou HBeAg-negativo, com doença hepática compensada ou descompensada.
- Estão disponíveis dados limitados de pacientes adultos com co-infecção HIV e VHB que tenham recebido terapia prévia com lamivudina.
Resultados de eficácia.
1-6
Desfechos na semana 48
A segurança e a eficácia de BARACLUDE foram avaliadas em três estudos Fase III de controle ativo. Esses estudos incluíram 1.633 pacientes com 16 anos de idade, ou mais, com infecção crônica por hepatite B (HBsAg-positivo sérico durante pelo menos 6 meses) acompanhada por evidência de replicação viral (DNA do VHB sérico detectável, medido pela hibridização do bDNA ou por ensaio PCR). Os pacientes apresentaram níveis de ALT persistentemente elevados ao menos 1,3 vezes o limite superior da normalidade (LSN) e inflamação crônica na biópsia do fígado, compatível com um diagnóstico de hepatite viral crônica. A segurança e a eficácia de BARACLUDE também foram avaliadas em um estudo de 191 pacientes com doença hepática descompensada infectados com VHB e em um estudo de 68 pacientes co-infectados com VHB e HIV.
Pacientes Virgens de Nucleosídeo com Doença Hepática Compensada
HBeAg-positivo: O AI 463022 foi um estudo multicêntrico, randomizado, duplo-cego de BARACLUDE 0,5 mg uma vez ao dia versus lamivudina 100 mg uma vez ao dia, por no mínimo 52 semanas, em 709 pacientes virgens de nucleosídeo (de 715 randomizados) com infecção crônica por hepatite B e HBeAg detectável. A idade média dos pacientes foi de 35 anos, 75% eram do sexo masculino; 57% eram asiáticos, 40% eram caucasianos e 13% haviam recebido anteriormente tratamento com interferon-a. No basal, os pacientes apresentaram uma média do Índice Necroinflamatório de Knodell de nível 7,8, de DNA de VHB sérico médio, avaliado através do ensaio Roche Amplicor® PCR, de 9,66 log10 cópias/mL e nível de ALT sérico médio de 143 U/L. Amostras de biópsia do fígado foram avaliadas em 89% dos pacientes.
HBeAg-negativo (anti-HBe positivo/DNA do VHB positivo): O AI 463027 foi um estudo multicêntrico, randomizado, duplo-cego de BARACLUDE 0,5 mg uma vez ao dia versus lamivudina 100 mg uma vez ao dia, por no mínimo 52 semanas em 638 pacientes virgens de nucleosídeo (de 648 randomizados), com infecção crônica por hepatite B HBeAg-negativo (HBeAb-positivo). A idade média dos pacientes foi de 44 anos e 76% eram do sexo masculino, 39% eram asiáticos e 58% eram caucasianos; 13% haviam recebido anteriormente tratamento com interferon-a. No basal, ospacientes apresentaram uma média do Índice Necroinflamatório de Knodell de nível 7,8 de DNA de VHB sérico médio, avaliado através do ensaio Roche Amplicor® PCR, de 7,58 log10 cópias/mL e um nível médio de ALT sérica de 142 U/L. Amostras de biópsia do fígado foram avaliadas em 88% dos pacientes.
Nos Estudos AI 463022 e AI 463027, o BARACLUDE foi superior à lamivudina no objetivo primário da eficácia de Melhora Histológica, definida como 2 pontos ou mais de redução no Índice Necroinflamatório de Knodell, sem piora no Índice de Fibrose de Knodell na semana 48 e no objetivo secundário de avaliações de eficácia de redução na carga viral e normalização ALT. A Melhora Histológica e a alteração no Índice de Fibrose de Ishak são mostradas na Tabela 1. Avaliações dos resultados bioquímicos, virológicos e sorológicos são mostradas na Tabela 2.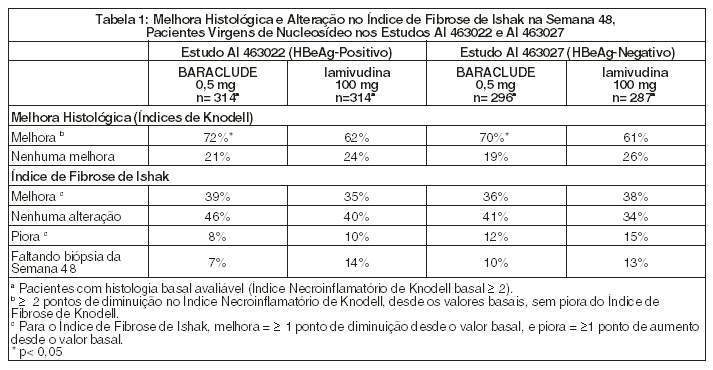
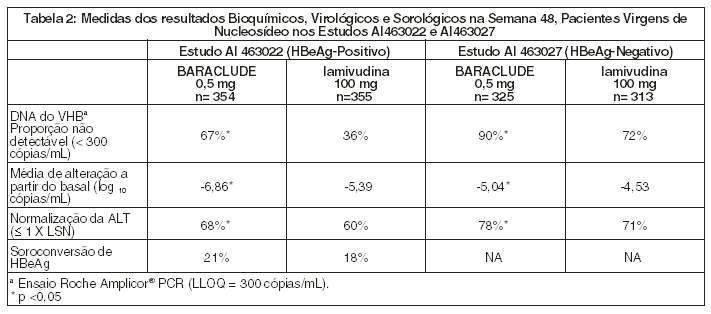
A melhora histológica foi independente dos níveis basais de DNA de VHB ou ALT.
Pacientes resistentes à lamivudina
O AI 463026 foi um estudo multicêntrico, randomizado, duplo-cego de BARACLUDE em 286 pacientes (de 293 randomizados) com infecção crônica por hepatite B resistente à lamivudina. Os pacientes que estavam recebendo lamivudina na admissão ao estudo trocaram pelo BARACLUDE 1 mg uma vez ao dia (sem um período de washout ou de sobreposição), ou continuaram com a lamivudina 100 mg por no mínimo 52 semanas. A idade média dos pacientes foi de 39 anos e 76% eram do sexo masculino; 37% eram asiáticos e 62% eram caucasianos e 52% haviam recebido anteriormente tratamento com interferon-a. A média de duração da terapia anterior com lamivudina foi 2,7 anos e 85% apresentavam mutações de resistência à lamivudina no basal em um exame investigacional. No basal, os pacientes apresentaram uma média de Índice Necroinflamatório de Knodell de nível 6,5, de DNA de VHB sérico médio, medido pelo ensaio Roche Amplicor® PCR, de 9,36 log10 cópias/mL e um nível sérico de ALT de 128 U/l. Amostras de biópsia do fígado foram coletadas em 87% dos pacientes.
BARACLUDE foi superior à lamivudina nas avaliações primárias de Melhora Histológica (utilizando o Índice de Knodell na Semana 48). Esses resultados e a alteração no índice de Fibrose de Ishak estão apresentados na Tabela 3. A Tabela 4 apresenta os resultados bioquímicos, virológicos e sorológicos.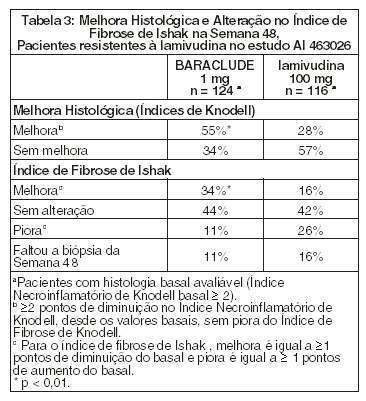
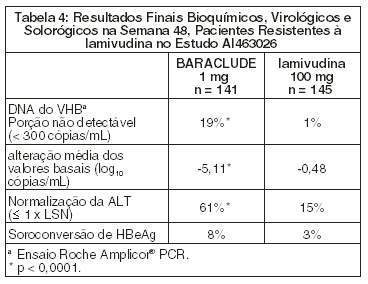
A melhora histológica foi independente dos níveis basais de DNA de VHB ou ALT.
Desfechos além da semana 48
A duração ideal de terapia com BARACLUDE é desconhecida. De acordo com os critérios dos protocolos dos estudos de fase III, os pacientes descontinuaram o tratamento com BARACLUDE ou lamivudina após 52 semanas de acordo com a definição de resposta baseada na supressão virológica do VHB ( < 0,7 MEq/mL por ensaio de bDNA) e perda de HBeAg (em pacientes HBeAg positivos) ou normalização de ALT < 1.25 X LSN (em pacientes HBeAg negativos) na semana 48. Pacientes que atingiram supressão virológica mas não resposta sorológica (HBeAg positivos) ou não atingiram ALT
< 1.25 X LSN (HBeAg negativos) continuaram recebendo dose cega por 96 semanas ou até a resposta ser atingida. Estes guias de gerenciamento específicos para pacientes de protocolo não são destinados como guias de práticas clínicas.
Pacientes virgens de tratamento com nucleosídeo: Entre pacientes HBeAg-positivo, virgens de tratamento (Estudo AI463022), 243 (69%) dos pacientes tratados com BARACLUDE e 164 (46%) tratados com lamivudina, continuaram em tratamento cego por até 96 semanas. Entre os pacientes que continuaram em tratamento cego no segundo ano, 180 (74%) e 60 (37%) dos pacientes tratados com BARACLUDE e lamivudina, respectivamente, atingiram DNA de VHB < 300 cópias/mL por PCR no final do tratamento (até 96 semanas). 193 (79%) dos pacientes tratados com BARACLUDE atingiram ALT ≤ X LSN, comparado a 112 (68%) dos pacientes tratados com lamivudina, e soroconversão de HBeAg ocorreu em 26 (11%) e em 20 (12%) dos pacientes tratados com BARACLUDE e lamivudina, respectivamente.
Entre pacientes HBeAg-positivo, virgens de tratamento, 74 (21%) dos que receberam terapia com BARACLUDE e 67 (19%) dos pacientes tratados com lamivudina, definiram resposta na 48° semana, descontinuaram o uso da droga e foram acompanhados sem tratamento por 24 semanas. Entre os responsivos ao BARACLUDE, 26 (35%) dos pacientes tiveram DNA de VHB < 300 cópias/mL, 55 (74%) tiveram ALT ≤ 1 X LSN e, 56 (76%) sustentaram soroconversão de HBeAg até o fim do acompanhamento. Entre os responsivos à lamivudina, 20 (30%) dos pacientes apresentaram DNA de VHB < 300 cópias/mL, 41 (61%) apresentaram ALT ≤ 1 X LSN, e 47 (70%) dos pacientes sustentaram soroconversão de HBeAg até o fim do acompanhamento.
Entre pacientes HBeAg-negativo (Estudo AI463027),virgens de tratamento, 26 (8%) e 28 (9%) dos pacientes tratados com BARACLUDE ou lamivudina, respectivamente, continuaram em tratamento cego por até 96 semanas. Neste pequeno tratamento contínuo de coorte no ano dois, 22 pacientes tratados com BARACLUDE e 16 com lamivudina, apresentaram DNA de VHB < 300 cópias/mL por PCR, e 7 e 6 pacientes, respectivamente, apresentaram ALT ≤ 1 X LSN no fim do tratamento (até 96 semanas).
Entre pacientes HBeAg-negativo, virgens de tratamento, 275 (85%) dos tratados com BARACLUDE e 245 (78%) dos tratados com lamividina definiram resposta na 48° semana, descontinuaram o tratamento e foram acompanhados sem tratamento por 24 semanas. Neste coorte, poucos pacientes em cada braço do tratamento tinham DNA de VHB < 300 cópias/mL por PCR no final do acompanhamento. No fim do acompanhamento, 126 (46%) dos pacientes tratados com BARACLUDE e 84 (34%) daqueles tratados com lamivudina apresentaram ALT ≤ 1 X LSN.
Pacientes resistentes à lamivudina: Entre os pacientes resistentes à lamivudina (Estudo AI463026), 77 (55%) dos tratados com BARACLUDE e 3 (2%) tratados com lamivudina continuaram em tratamento cego por até 96 semanas. Neste coorte de pacientes tratados com BARACLUDE, 31 (40%) atingiram DNA de VHB < 300 cópias/mL, 62 (81%) dos pacientes tiveram ALT ≤ 1 X LSN, e 8 (10%) demonstraram soroconversão de HBeAg no fim do tratamento.
Populações Especiais
-Pacientes com doença hepática descompensada
Em um estudo randomizado aberto, 191 pacientes com infecção crônica por VHB com HBeAg-positivo ou negativo, com evidência de descompensação hepática, definida como um pontuação de Child Turcotte-Pugh (CTP) de 7 ou maior, receberam BARACLUDE 1 mg uma vez ao dia ou adefovir dipivoxil 10 mg uma vez ao dia. Os pacientes ou eram virgens de tratamento para VHB ou préviamente tratados (excluindo pré tratamento com BARACLUDE, adefovir dipivoxil ou tenofovir disoproxil fumarato). BARACLUDE foi superior ao adefovir dipivoxil no desfecho de eficácia primário de alteração média do valor basal no DNA de VHB sérico por PCR, na semana 24. Resultados para os desfechos selecionados do estudo, na semana 24 e 48, são apresentados na tabela 5.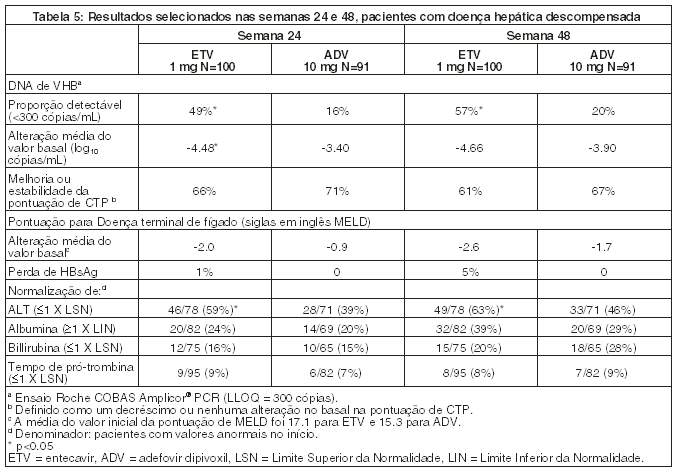
O tempo para início de carcinoma hepatocelular ou morte (qualquer um que ocorra primeiro) foi comparável nos dois grupos de tratamento.
-Pacientes co-infectados com HIV e VHB
O AI 463038 foi um estudo randomizado, duplo-cego, controlado com placebo de BARACLUDE versus placebo, em 68 pacientes co-infectados com HIV e VHB, que apresentaram recorrência de viremia de VHB enquanto recebiam um regime HAART (highly active antiretroviral therapy) [terapia anti-retroviral altamente ativa] contendo lamivudina. Os pacientes continuaram com seu regime HAART contendo lamivudina (300 mg/dia) e foram designados para acrescentar 1 mg de BARACLUDE uma vez ao dia (51 pacientes) ou placebo (17 pacientes), durante 24 semanas, seguidas por uma fase aberta, por um período adicional de 24 semanas, em que todos os pacientes receberam BARACLUDE. No basal, os pacientes apresentaram um nível sérico médio de DNA do VHB, por PCR, de 9,13 log10 cópias/mL. 99% dos pacientes eram HBeAg-positivos no basal, com um nível basal médio de ALT de 71,5 U/l. Os níveis médios de RNA HIV permaneceram estáveis em aproximadamente 2 log 10 cópias/mL até a semana 24 da terapia cega. Os resultados bioquímicos e virológicos na semana 24 são apresentados na Tabela 6. Não há dados em pacientes co-infectados com HIV e VHB, que não receberam terapia prévia com lamivudina. BARACLUDE não foi avaliado em pacientes co-infectados com HIV e VHB que não estivessem recebendo simultaneamente tratamento efetivo para HIV.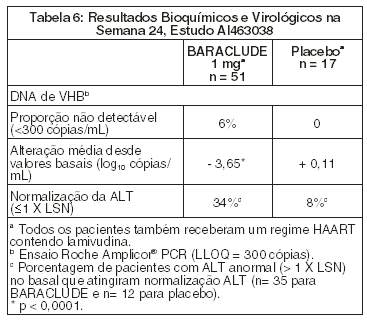
Para pacientes originalmente determinados para BARACLUDE, no fim da fase aberta (semana 48), 8% dos pacientes tinham DNA de VHB < 300 cópias/mL por PCR, a principal alteração do basal de DNA de VHB por PCR foi de - 4,20 log10 cópias/mL, e 37% dos pacientes com ALT anormal no basal tiveram normalização ALT ( < 1 X LSN).
Caract. farmacológicas.
BARACLUDE é um análogo de nucleosídeo guanosino, com atividade seletiva contra o vírus da hepatite B (VHB). O nome químico do entecavir é 2-amino-1,9-diidro-9-[(1S,3R,4S)-4-hidróxi-3-(hidroximetil)-2metileneciclopentil]-6H-purina-6-um, monoidratado. Sua fórmula molecular é C12H15N5O3•H2O, que corresponde a um peso molecular de 295,3. O entecavir apresenta a seguinte fórmula estrutural: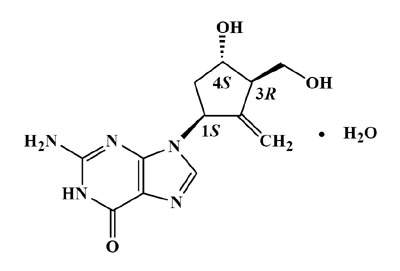
Farmacodinâmica
Mecanismo de Ação
O entecavir é um análogo de nucleosídeo guanosino, com uma atividade contra a polimerase do VHB. Ele é eficientemente fosforilado na forma ativa de trifosfato (TP), que tem uma meia-vida intracelular de 15 horas. Através da competição com o substrato natural deoxiguanosina TP, entecavir-TP inibe funcionalmente todas as três atividades da polimerase viral (transcriptase reversa, rt): (1) atividade principal da polimerase VHB, (2) transcrição reversa da fita negativa do mensageiro pregenômico do RNA, e (3) síntese da fita positiva de DNA do VHB. Entecavir-TP é um inibidor fraco das polimerases de DNA celular a, b, e d e da polimerase de DNA mitocondrial c com valores de Ki variando de 18 a > 160mM.
Atividade antiviral
A concentração de entecavir que inibiu 50% da síntese do DNA viral (EC50) foi de 0,004 mm em células humanas HepG2 transfectadas com o tipo selvagem de VHB. O valor médio de EC50 para entecavir contra VHB resistentes à lamivudina (rtL 180M, rtM204V) foi de 0,026mM (entre 0,010 -0,059mM).
A coadministração de inibidores da transcriptase reversa de nucleosídeo (ITRNs) de HIV com o BARACLUDE não tem a probabilidade de reduzir a eficácia antiviral do BARACLUDE contra o VHB, ou de nenhum desses agentes contra o HIV. Em ensaios de combinação do VHB in vitro, a atividade antiviral do entecavir, não foi afetada pela presença de abacavir, didanosina, lamivudina, estavudina, tenofovir ou zidovudina. Em ensaios antivirais do HIV, o entecavir não apresentou efeito sobre a atividade anti-HIV in vitro destes seis ITRNs ou emtricitabina em concentrações > 100 vezes a Cmax de entecavir com 1mg de dose.
Atividade antiviral contra o HIV
Uma análise abrangente da atividade inibitória do entecavir em isolados laboratoriais e clínicos do HIV tipo 1 (HIV-1), utilizando-se uma variedade de células e condições de ensaio, resultou em valores de EC50 que variaram de 0,026 a > 10 mM. Os valores mais baixos de EC50 foram observados quando houve diminuição da carga viral utilizada no ensaio. Em cultura de células, o entecavir selecionou para uma substituição M184I na transcriptase reversa do HIV, em concentrações micromolares, confirmando sua pressão inibitória em concentrações elevadas. Variantes do HIV, com substituição M184V, apresentaram perda de sensibilidade ao entecavir.
Resistência à droga
- In vitro
Em ensaios baseados em células, reduções de 8 a 30 vezes na sensibilidade fenotípica do entecavir foram observadas para as cepas resistentes à lamivudina. Para reduções adicionais ( > 70 vezes) na sensibilidade fenotípica do entecavir, foi necessária a presença de substituições aminoácidas no rtM204I/V com ou sem alteração em rtL180M, e substituições adicionais nos resíduos rtT184, rtS202, ou rtM250 ou a combinação destas substituições com ou sem uma substituição rtI169 na polimerase do VHB.
- Estudos Clínicos
Pacientes virgens de tratamento com nucleosídeo: Avaliações genotípicas foram realizadas em amostras ( > 300 cópias/mL de soro com DNA de VHB) de 562 pacientes tratados com BARACLUDE por até 96 semanas em estudos com pacientes virgens de tratamento com nucleosídeo (AI463022, AI463027, e estudo de extensão AI463901). Na semana 96, a evidência de substituição de aminoácido na rtS202G, com substituições na rtM204V e rtL180M, foi detectada em VHB de 2 pacientes (2/562 = < 1%), e um deles apresentou rebote virológico (≥1 log de aumento sobre nadir). Além disso, substituições de aminoácido na rtM204I/V e rtL180M, rtL80I, ou rtV173L, que levaram a diminuição da sensibilidade fenotípica ao entecavir na ausência de mudanças na rtT184, rtS202, ou rtM250, foram detectados no VHB de 3 pacientes (3 / 562 = < 1%) que apresentaram rebote virológico. Para pacientes que continuaram o tratamento após 48 semanas, 75% (202/269) tiveram DNA de VHB < 300 cópias / mL no final do tratamento (até 96 semanas).
Pacientes virgens de tratamento HBeAg-positivo (n=243) e HBeAg-negativo (n=39), que falharam para atingir a resposta completa definida do estudo em 96 semanas, continuaram o tratamento com entecavir em um estudo de extensão. A resposta completa para HbeAg-positivo foi < 0.7 MEq/mL (aproximadamente 7 x 105 cópias/mL) de DNA de VHB sérico e perda de HBeAg e, para HbeAgnegativo foi < 0.7 MEq/mL de DNA de VHB e normalização ALT. Pacientes receberam 1 mg de entecavir uma vez ao dia por um período adicional de até 144 semanas. Desses 282 pacientes, 141 pacientes HBeAg - positivo e 8 pacientes HBeAg -negativo entraram no estudo de extensão de acompanhamento de longa duração e foram avaliados para resistência ao entecavir. Entre os 149 pacientes que entraram no estudo extensão, 88% (131/149), 92% (137/149) e 92% (137/149) atingiram DNA de VHB sérico < 300 cópias/mL nas semanas 144, 192 e 240 (incluindo final do tratamento), respectivamente. Nenhuma nova substituição associada à resistência ao entecavir foi identificada na comparação dos genótipos dos isolados avaliados com seus respectivos isolados basais. A probabilidade cumulativa de desenvolvimento das substituições relacionadas à resistência ao entecavir em rtT184, rtS202, ou rtM250 (na presença das substituições em rtM204V e rtL180M) nas semanas 48, 96, 144, 192 e 240 foi 0.2%, 0.5%, 1.2%, 1.2% e 1.2%, respectivamente.
Pacientes resistentes à lamivudina: Avaliações genotípicas foram realizadas em amostras disponíveis de 190 pacientes tratados com BARACLUDE por até 96 semanas em estudos de VHB resistente a lamivudina (AI463026, AI463014, AI463015, e estudo de extensão AI463901). Na semana 96, substituições de aminoácidos associadas à resistência em rtS202, rtT184, -ou rtM250, com ou sem alterações em rtI169, na presença de substituições aminoácidas em rtM204I/V com ou sem rtL180M, rtL80V ou rtV173L/M surgiram no VHB de 22 pacientes (22/190 = 12%), sendo que 16 apresentaram rebote virológico (≥ 1 log de aumento sobre nadir) e 4 nunca tiveram supressão viral < 300 cópias/mL. O VHB de 4 desses pacientes possuiam substituições de resistência ao entecavir no basal e apresentaram alterações no tratamento com entecavir. Além dos 22 pacientes, 3 outros apresentaram rebote virológico com o surgimento de rtM204I/V e rtL180M, rtL80V, ou rtV173L/M. Para isolados de pacientes que apresentaram rebote virológico com o aparecimento de substituições de resistência (n=19), a média de alteração nos valores de EC50 do entecavir quando comparado à referência, foi 19 vezes no basal e 106 vezes no período de rebote virológico. Para pacientes que continuaram o tratamento além das 48 semanas, 40% (31/77) tinham DNA de VHB < 300cópias/mL no final do tratamento (até 96 semanas)
Pacientes resistentes à lamivudina (n=157), que falharam para atingir resposta completa definida do estudo na semana 96, continuaram o tratamento com entecavir. Os pacientes receberam 1mg de entecavir uma vez ao dia por um período adicional de até 144 semanas. Desses pacientes, 80 entraram no estudo de acompanhamento de longa duração e foram avaliados para a resistência ao entecavir. Nas semanas 144, 192 e 240 (incluindo o final do tratamento), 34% (27/80), 35% (28/80), e 36% (29/80), respectivamente, atingiram DNA de VHB < 300 cópias/mL. A probabilidade cumulativa de desenvolvimento de substituições rtT184, rtS202, ou rtM250 associadas à resistência ao entecavir (na presença de substituições rtM204I/V com ou sem rtL180M) nas semanas 48, 96, 144, 192 e 240 foi 6.2%, 15%, 36.3%, 46.6% e 51.5%, respectivamente. O VHB de 6 pacientes desenvolveram substituições de aminoácidos na rtA181C/G/S/T enquanto eram tratados com entecavir, desses, 4 desenvolveram substituições de resistência ao entecavir nas rtT184, rtS202, ou rtM250 e um tinha uma substituição na rtT184S no basal. Dos 7 pacientes cujo VHB tinha uma substituição na rtA181 no basal, 2 pacientes também tinham substituições na rtT184, rtS202, ou rtM250 no basal e outros 2 pacientes desenvolveram-nas enquanto estavam sob tratamento com entecavir.
Resistência Cruzada: Resistência cruzada foi observada entre análogos de nucleosídeos de VHB. Em ensaios baseados em células, entecavir apresentou inibição de 8 à 30 vezes menor na síntese do DNA de VHB para o VHB contendo substituições de resistência à lamivudina e telbivudina rtM204I/V com ou sem rtL180M em relação ao vírus selvagem de VHB. Substituições rtM204I/V com ou sem rtL180M, rtL80I/V,ou rtV173L, que estão associadas com resistência à lamivudina e telbivudina, também decrescem a sensibilidade fenotípica ao entecavir. A eficácia de entecavir contra VHB com substituições associadas à resistência ao adefovir não foram estabelecidas nos estudos clínicos. Isolados de VHB de pacientes resistentes à lamivudina com falha na terapia com entecavir, foram susceptíveis ao adefovir em cultura celular mas permaneceram resistentes à lamivudina. Genomas recombinantes de VHB codificando substituições associadas à resistência ao adefovir em rtN236T ou rtA181V, apresentaram 0,3 e 1,1 vezes de alteração na susceptibilidade ao entecavir em cultura celular, respectivamente.
-Farmacocinética
As farmacocinéticas de dose única e múltipla de entecavir foram avaliadas em voluntários sadios e pacientes com infecção crônica de hepatite B.
-Absorção
Em voluntários sadios, entecavir foi rapidamente absorvido com pico de concentração plasmática ocorrendo entre 0,5 e 1,5 horas após administração oral. Houve um aumento proporcional a dose nos valores de pico de concentração plasmática (Cmáx) e área sobre a curva (AUC) no estado de equilíbrio de acordo com doses múltiplas diárias entre 0,1 e 1mg. O estado de equilíbrio foi atingido após 6-10 dias da administração de uma dose única diária com acúmulo aproximado de 2 vezes a concentração. Cmáx e concentração plasmática no estado de equilíbrio foram de 4,2 e 0,3 ng/mL, respectivamente, para a dose de 0,5 mg, e 8,2 e 0,5 ng/mL, respectivamente, para dose de 1mg.
Efeitos da alimentação na absorção oral: A administração oral de entecavir 0,5mg com refeição padrão gordurosa (945kcal, 54,6g de gordura) ou refeição leve (379kcal, 8,2g de gordura) resultou em atraso na absorção (1-1,5 hora alimentado vs. 0,75 hora em jejum), diminuição no Cmáx de 44-46%, e diminuição no AUC de 18-20%.
- Distribuição
Baseando-se no perfil farmacocinético de entecavir após administração oral, o volume aparente de distribuição estimado para entecavir, está em excesso no total de água presente no corpo, sugerindo que entecavir é distribuído extensivamente nos tecidos.
A ligação de entecavir às proteínas plasmáticas humanas in vitro, foi aproximadamente 13%.
-Metabolismo e eliminação
Após a administração de 14C-entecavir em humanos e ratos, nenhum metabólito oxidativo ou acetilado foi observado. Quantidade mínima de metabólitos de fase II (conjugados de glucuronida e sulfato) foram observados.
Entecavir não é um substrato, inibidor ou indutor do sistema enzimático CYP450. (vide 3. CARACTERÍSTICAS FARMACOLÓGICAS: Interações medicamentosas)
Após atingir níveis de pico, as concentrações plasmáticas de entecavir diminuíram de uma maneira biexponencial com meia-vida de eliminação terminal de aproximadamente 128-149 horas. O índice de acúmulo de droga observado é aproximadamente 2 vezes maior com dose única diária, sugerindo um tempo de meia-vida efetivo de aproximadamente 24 horas.
Entecavir é predominantemente eliminado pelos rins com recuperação urinária de droga inalterada no estado de equilíbrio de 62%-73% da dose administrada. O clearance renal é independente da dose e varia entre 360 e 471mL/min sugerindo que entecavir passa tanto por filtração glomerular quanto por secreção da rede tubular. (vide 6. INTERAÇÕES MEDICAMENTOSAS)
- Populações Especiais
Pacientes com Insuficiência Renal
A farmacocinética do entecavir após uma dose única de 1 mg foi estudada em pacientes (sem infecção crônica por hepatite B) com graus selecionados de insuficiência renal, incluindo pacientes cuja insuficiência renal era controlada por hemodiálise ou por diálise peritoneal ambulatorial contínua (DPAC). Os resultados são apresentados na Tabela 7: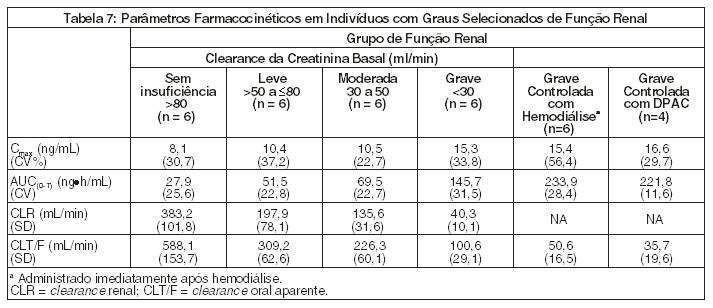
Após uma dose única de 1 mg de entecavir administrado 2 horas antes da sessão de hemodiálise, foi removida aproximadamente 13% da dose de entecavir durante 4 horas. DPAC removeu aproximadamente 0,3% da dose, durante 7 dias.
Pacientes com Insuficiência Hepática
A farmacocinética do entecavir após uma dose única de 1 mg foi estudada em pacientes (sem infecção crônica por hepatite B) com insuficiência hepática moderada e grave (Child-Pugh classe B ou C). A farmacocinética do entecavir foi semelhante entre os pacientes com insuficiência hepática e indivíduos saudáveis de controle; portanto, nenhum ajuste na dosagem de BARACLUDE é recomendado para pacientes com insuficiência hepática.
Pacientes que Receberam Transplante de Fígado
A segurança e eficácia de BARACLUDE em pacientes que receberam transplante de fígado são desconhecidas. Porém, em um pequeno estudo piloto de uso de entecavir em receptores de transplante de fígado, infectados por VHB, com uma dose estável de ciclosporina A (n=5) ou de tacrolimus (n = 4), a exposição ao entecavir foi de aproximadamente 2 vezes a exposição em indivíduos saudáveis, com função renal normal. A função renal alterada contribuiu para o aumento da exposição ao entecavir nesses pacientes. O potencial de interações farmacocinéticas entre entecavir e ciclosporina A ou tacrolimus não foi formalmente avaliado.
Pacientes Geriátricos
O efeito da idade na farmacocinética do entecavir foi avaliado através da administração de uma dose única oral de 1mg em voluntários jovens e idosos sadios. O AUC de entecavir foi de 29,3% maior em voluntários idosos em relação aos jovens. A disparidade na exposição entre voluntários jovens e idosos foi provavelmente atribuída a diferenças na função renal. O ajuste de dose de BARACLUDE deve ser baseado na função renal do paciente e não na idade.
Sexo/Raça
Sexo ou raça não interfere no perfil farmacocinético de entecavir.
- Interações medicamentosas
O metabolismo de entecavir foi avaliado em estudos in vitro e in vivo. Entecavir não é um substrato, inibidor ou indutor do sistema enzimático citocromo P450 (CYP450). Em concentrações de até aproximadamente 10.000 vezes maiores que aquelas obtidas em humanos, entecavir não inibiu nenhuma das principais enzimas do CYP450 1A2, 2C9, 2C19, 2D6, 3A4, 2B6 e 2E1. Em concentrações de até aproximadamente 340 vezes maiores que aquelas observadas em humanos, entecavir não induziu as enzimas humanas do CYP450 1A2, 2C9, 2C19, 3A4, 3A5 e 2B6.
É improvável que a farmacocinética do entecavir seja afetada pela co-administração de agentes que também sejam metabolizados pelo sistema CYP450, induzem ou inibem o sistema CYP450. Da mesma maneira, é improvável que a farmacocinética de conhecidos substratos de CYP seja afetada pela coadministração de entecavir.
O estado de equilíbrio farmacocinético de entecavir e drogas coadministradas, não foi alterado em estudos de interação de entecavir com lamivudina, adefovir dipivoxil e tenofovir disoproxil fumarato (vide 6. INTERAÇÕES MEDICAMENTOSAS).
Contraindicações.
BARACLUDE é contraindicado em pacientes que previamente demonstraram hipersensibilidade ao entecavir ou a qualquer outro componente do produto.
Advertências e precauções.
Atenção: o tratamento com BARACLUDE não diminui o risco de transmissão do VHB para outras pessoas através do sexo, compartilhamento de agulhas ou exposição ao sangue. Converse com seu paciente sobre a prática de sexo seguro. Oriente-o para nunca compartilhar agulhas ou itens pessoais que possam conter sangue ou fluídos corpóreos, como escovas de dente ou lâminas de barbear.
Exacerbação aguda grave da hepatite B
Exacerbação aguda de hepatite B foi relatada em pacientes que descontinuaram terapia para hepatite B, incluindo a terapia com entecavir. A maioria das exacerbações pós-tratamento parecem ser autolimitadas. No entanto, exacerbações graves, incluindo casos fatais, podem ocorrer. A relação causal desses eventos com a descontinuação da terapia é desconhecida. A função hepática deve ser monitorada com acompanhamento clínico e laboratorial, por alguns meses, em pacientes que descontinuaram a terapia para hepatite B. Se apropriada, a reintrodução da terapia para hepatite B deve ser autorizada. (vide 9. REAÇÕES ADVERSAS: Exacerbações da hepatite após Descontinuação do Tratamento).
Pacientes Co-infectados com HIV e VHB
BARACLUDE não foi avaliado em pacientes co-infectados com HIV e VHB que não estejam recebendo simultaneamente tratamento efetivo para HIV. Estudos clínicos limitados sugerem que existe um potencial para o desenvolvimento da resistência aos inibidores da transcriptase reversa de nucleosídeo (ITRNs) de HIV se BARACLUDE for usado no tratamento de hepatite B crônica em pacientes com HIV que não estão sendo tratados. Portanto, a terapia com BARACLUDE não é recomendada em pacientes co-infectados que não estão recebendo terapia ativa antiretroviral (HAART). Antes de iniciar a terapia com BARACLUDE, os pacientes devem realizar um teste para HIV. BARACLUDE não foi estudado para tratamento de pacientes com HIV e não é recomendado para para este uso.
Acidose láctica e hepatomegalia grave com esteatose
Acidose láctica e hepatomegalia grave com esteatose, incluindo casos fatais, foram relatados com o uso de análogos de nucleosídeos isolados ou em combinação com antiretrovirais.
A maioria dos casos tem ocorrido em mulheres. Obesidade e prolongamento da exposição aos nucleosídeos podem ser fatores de risco. Deve-se ter cautela na administração de análogos de nucleosídeos em qualquer paciente com fator de risco conhecido para doença de fígado, entretanto, alguns casos tem sido reportados em pacientes sem histórico desses fatores de risco. O tratamento com BARACLUDE deverá ser suspenso em qualquer paciente que apresentar sintomas clínicos ou achados laboratoriais sugestivos de acidose láctica ou hepatotoxicidade pronunciada (a qual pode incluir hepatomegalia e esteatose mesmo na ausência de elevações de transaminases acentuadas).
Carcinogênese, Mutagênese, e Infertilidade
Estudos de carcinogenicidade oral a longo prazo em camundongos e ratos foram conduzidos com exposições de até 42 vezes (camundongos) e 35 vezes (ratos) daquelas observadas em humanos, com maior dose recomendada de 1 mg/dia. Em estudos com camundongos e ratos, entecavir foi positivo para achados carcinogênicos.
Em camundongos, adenomas de pulmão aumentaram em machos e fêmeas com exposição de 3 a 40 vezes daquelas em humanos. Carcinomas de pulmão aumentaram em camundongos machos e fêmeas com exposição de 40 vezes daquelas em humanos. Adenomas e carcinomas combinados aumentaram em camundongos machos com exposição 3 vezes e em camundongos fêmeas com exposição 40 vezes daquelas em humanos. O desenvolvimento de tumor foi precedido por proliferação de pneumócitos nos pulmões, o que não foi observado em ratos, cães ou macacos aos quais foi administrado o entecavir, dando suporte à conclusão de que os tumores nos pulmões de camundongos são um evento específico da espécie. Carcinomas hepatocelulares aumentaram em machos e adenomas combinado com carcinoma hepático também aumentaram com exposição 42 vezes daquelas em humanos. Tumores vasculares em camundongos fêmeas (hemangiomas de ovários e útero e hemangiosarcomas de baço) aumentaram com exposição de 40 vezes daquelas em humanos. Em ratos, adenomas hepatocelulares aumentaram em fêmeas com exposição de 24 vezes daquelas em humanos; carcinomas combinados com adenomas também aumentaram em fêmeas com exposição de 24 vezes daquelas em humanos. Gliomas cerebrais foram induzidos em machos e fêmeas com exposição de 35 a 24 vezes daquelas em humanos. Fibromas de pele foram induzidos em fêmeas com exposição de 4 vezes daquelas em humanos.
Não se sabe se os resultados dos estudos de carcinogenicidade em roedores são preditivos para humanos.
O entecavir foi clastogênico para culturas de linfócito humano. Entecavir não foi mutagênico no ensaio de mutagenicidade microbiana Ames, usando culturas de S. typhimurium e E. coli na presença ou ausência de ativação metabólica, um ensaio de mutação de gene em célula de mamíferos e um ensaio de transformação, com células de embrião de hamster sírio. O entecavir também foi negativo em um estudo oral de micronúcleo e em um estudo oral de reparo de DNA em ratos.
Em estudos de toxicidade reprodutiva, em que foram administrados nos animais doses de até 30mg/kg por até 4 semanas, não houve evidência de efeitos sobre a fertilidade em ratos machos e fêmeas com exposição sistêmica > 90 vezes aquelas atingidas em humanos com maior dose recomendada de 1 mg/dia. Em estudos de toxicidade em roedores e cães, foi observada uma degeneração tubular seminífera com exposições > 35 vezes daquelas atingidas em humanos. Nenhuma alteração testicular foi evidente em macacos.
Uso em populações específicas
-Gravidez
Categoria de risco na gravidez - C
Este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica.
Não existem estudos adequados e bem controlados em mulheres grávidas. Considerando que estudos de reprodução não são sempre preditivos para a resposta humana, BARACLUDE deve ser utilizado durante a gravidez somente se realmente necessário e após consideração cuidadosa dos riscos e benefícios. Estudos de reprodução foram realizados em ratos e coelhos. Não houve sinais de toxicidade embriofetal ou materna quando animais prenhas receberam entecavir em exposições de 28 (ratos) e 212 (coelhos) vezes aproximadamente da exposição humana atingida na maior dose recomendada em humanos de 1 mg/dia. Em ratos, toxicidade materna, toxicidade embriofetal (reabsorções), pesos corporais fetais mais baixos, malformações vertebrais da cauda, ossificação reduzida (vértebras, esternebras e falanges), vértebras extra-lombares e costelas foram observadas em exposições de 3100 vezes daquelas em humanos. Em coelhos, toxicidade embriofetal (reabsorções), ossificação reduzida (hióide) e uma incidência aumentada da 13ª costela foram observadas em exposições de 883 vezes daquelas em humanos. Em um estudo pré-natal e pós-natal em ratos, não foi observado nenhum efeito adverso sobre a descendência com administração oral de entecavir com exposições > 94 vezes à exposição humana.
-Trabalho de Parto
Não há estudos em mulheres grávidas e não há dados sobre o efeito do BARACLUDE sobre a transmissão do VHB de mãe para filho. Portanto, intervenções apropriadas devem ser utilizadas para evitar a aquisição neonatal do VHB.
-Lactação
O entecavir é excretado no leite de ratas. Não se sabe se essa droga é excretada no leite humano. Mães devem ser instruídas a não amamentar, caso estejam tomando BARACLUDE.
- Uso Pediátrico
A segurança e eficácia de entecavir em pacientes com menos de 16 anos de idade não foram estabelecidas.
- Uso Geriátrico
Os estudos clínicos de BARACLUDE não incluem número suficiente de indivíduos com 65 anos ou mais, para determinar se eles respondem de maneira diferente aos indivíduos mais jovens. O entecavir é excretado principalmente pelos rins, o risco de reações tóxicas a essa droga pode ser maior em pacientes com função renal prejudicada. Como os pacientes idosos têm uma probabilidade maior de apresentar função renal diminuída, deve-se tomar cuidado na escolha da dose, e pode ser interessante monitorar a função renal (vide 8. POSOLOGIA E MODO DE USAR: Pacientes com Insuficiência Renal).
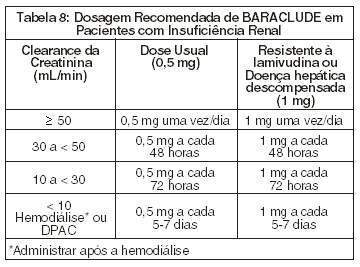
-Pacientes com Insuficiência Renal
O ajuste de dose de BARACLUDE é recomendado para pacientes com clearance de creatinina < 50mL/min, incluindo pacientes sob hemodiálise ou diálise peritoneal ambulatorial contínua (DPAC).
Pacientes que Receberam Transplante de Fígado
A segurança e eficácia de BARACLUDE em pacientes que receberam transplante de fígado são desconhecidas. Se o tratamento com BARACLUDE for necessário em pacientes que receberam transplante de fígado e que receberam ou estão recebendo imunossupressores que podem afetar a função renal, como ciclosporina ou tacrolimus, a função renal deve ser cuidadosamente avaliada antes e durante o tratamento com BARACLUDE.
-Uso em grupos raciais/étnicos
Estudos clínicos de BARACLUDE não incluíram número suficiente de pacientes de algumas raças/etnias minoritárias (americanos negros/africanos, hispânicos) para determinar se eles respondem de forma diferente ao tratamento com a droga. Não existe diferenças significativas, relacionadas com a raça, na farmacocinética do entecavir.
Interações medicamentosas.
Como entecavir é predominantemente eliminado pelos rins, a co-administração de BARACLUDE com medicamentos que reduzem a função renal ou que competem por secreção tubular ativa pode aumentar as concentrações dos dois medicamentos no sangue.
A co-administração de entecavir com lamivudina, adefovir dipivoxil ou tenofovir disoproxil fumarato resultou em interações medicamentosas não significantes. Os efeitos da co-administração de entecavir com outros medicamentos que são excretados pelos rins ou que afetam a função renal não foram avaliados. Pacientes devem ser cuidadosamente monitorados quanto a eventos adversos, quando BARACLUDE é co-administrado com estes medicamentos.
Interação com alimentos
A administração de entecavir com alimentos reduz a absorção (vid