AVSOLA
AMGEN
infliximabe
Anticorpo monoclonal.
Apresentações.
AVSOLA 100mg pó liofilizado para solução injetável: cada embalagem contém um frasco-ampola com 100 mg de infliximabe para reconstituição em 10 mL de água para injetáveis, e posteriormente diluído em cloreto de sódio 0,9% para infusão.
USO INTRAVENOSO
USO ADULTO E PEDIÁTRICO ACIMA DE 6 ANOS
Composição.
Cada frasco-ampola contém: infliximabe 10 mg/mL. Excipientes: sacarose, fosfato de sódio dibásico anidro, fosfato de sódio monobásico monoidratado, polissorbato 80 q.s.
Informações técnicas.
1. INDICAÇÕES
Artrite reumatoide
AVSOLA é uma "Terapia Antirreumática Controladora da Doença" (DCART - Disease-Controlling Anti-Rheumatic Therapy) indicado para:
- redução de sinais e sintomas;
- prevenção de lesão articular estrutural (erosões e estreitamento do espaço articular)
- melhora na função física;
em pacientes com doença ativa já tratados com metotrexato (artrite reumatoide estabelecida) e com doença ativa ainda não tratados com metotrexato (artrite reumatoide inicial).
Espondilite anquilosante
AVSOLA é indicado para:
- redução dos sinais e sintomas;
- melhora na função física;
em pacientes com doença ativa.
Artrite psoriásica
AVSOLA é indicado para:
Tratamento da artrite psoriásica ativa e progressiva em adultos, que tiveram resposta inadequada às drogas modificadoras da doença (DMARDs).
AVSOLA deve ser administrado:
- em associação com metotrexato;
- ou em monoterapia em pacientes que demonstraram intolerância ao metotrexato ou aqueles para os quais o metotrexato é contraindicado.
Infliximabe demonstrou melhorar a função física e inibir a progressão da lesão estrutural da artrite ativa, de acordo com a avaliação de radiografias dos pacientes com artrite psoriásica.
Psoríase em placa
AVSOLA é indicado para:
- redução dos sinais e sintomas da psoríase em placa;
- melhora na qualidade de vida;
no tratamento de pacientes adultos com psoríase em placa grave candidatos à terapia sistêmica, e para aqueles com psoríase em placa moderada em que a fototerapia é inadequada ou imprópria.
Doença de Crohn adulto e pediátrico
AVSOLA é indicado para:
- redução de sinais e sintomas;
- indução e manutenção da remissão clínica;
- indução da cicatrização da mucosa em adultos;
- melhora na qualidade de vida;
em pacientes com doença de Crohn de moderada a grave que tiveram uma resposta inadequada às terapias convencionais. A terapia com AVSOLA permite a redução ou suspensão do uso de corticosteroides pelos pacientes.
Doença de Crohn fistulizante
AVSOLA é indicado para:
- redução no número de fístulas enterocutâneas com drenagem e fístulas retovaginais e manutenção da fístula cicatrizada;
- redução dos sinais e sintomas;
- melhora na qualidade de vida;
em pacientes com doença de Crohn fistulizante.
Colite ou retocolite ulcerativa adulto e pediátrico
AVSOLA é indicado para:
- redução dos sinais e sintomas;
- indução e manutenção da remissão clínica;
- indução e manutenção da cicatrização da mucosa;
- melhora na qualidade de vida em adultos;
- redução ou descontinuação do uso de corticosteroides;
- redução da hospitalização relacionada à colite ou retocolite ulcerativa em adultos;
- redução da incidência de colectomia em adultos; em pacientes com colite ou retocolite ulcerativa ativa com resposta inadequada aos tratamentos convencionais. AVSOLA é também indicado para a redução da incidência de colectomia em pacientes adultos com colite ou retocolite ulcerativa moderada ou gravemente ativa, refratária a corticosteroides intravenosos.
2. RESULTADOS DE EFICÁCIA
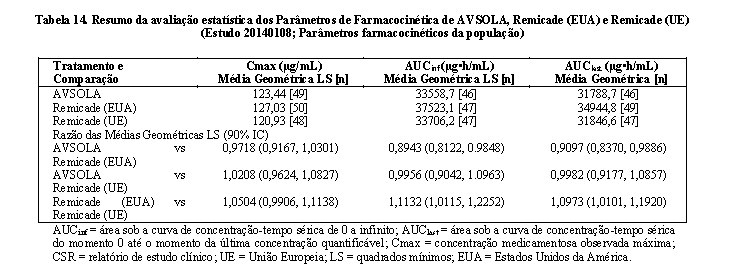
AVSOLA é um medicamento biológico desenvolvido pela via da comparabilidade (biossimilar). O programa de desenvolvimento clinico do produto foi projetado para demonstrar a comparabilidade entre AVSOLA e o produto comparador Remicade (infliximabe).
Dados de eficácia de AVSOLA
Estudo Comparativo entre AVSOLA e Remicade (Estudo 20140111)
O estudo 20140111 foi um estudo randomizado, duplo-cego, controle ativo em pacientes adultos com artrite reumatoide moderada a severa que tiveram resposta inadequada ao metotrexato (MTX). O estudo foi desenhado para comparar eficácia, segurança, imunogenicidade e farmacocinética de AVSOLA com Remicade (infliximabe).
Os pacientes elegíveis tiveram o diagnóstico de artrite reumatoide ativa durante pelo menos 3 meses definido como pelo menos 6 articulações inchadas, 6 articulações sensíveis e elevada taxa de sedimentação de eritrócitos [ESR ( > 28 mm/hr)] ou proteína C reativa sérica [CRP ( > 1,0 mg/ dL)] no início do estudo. Os indivíduos elegíveis também apresentavam fator reumatoide positivo e/ou anti-peptídeo citrulinado cíclico no momento da triagem, tinham tomado MTX por ≥ 12 semanas consecutivas e estavam com uma dose estável de MTX (7,5 a 25 mg/semana) por ≥ 8 semanas.
Para AVSOLA e Remicade, a média de idade foi respectivamente 55 e 54,8 anos, 76,7% e 79,9% foram mulheres, e 27,6% e 29,0% utilizaram biológicos para artrite reumatoide anteriormente.
Um total de 558 pacientes foram randomizados na proporção 1:1 para o recebimento da dose de 3mg/kg IV de AVSOLA ou de Remicade no dia 1, nas semanas 2 e 6, e a cada 8 semanas até a semana 22. Na semana 22, os pacientes que foram inicialmente randomizados para Remicade foram re-randomizados na proporção 1:1 para continuar recebendo Remicade a cada 8 semanas (referido como grupo de tratamento Remicade/Remicade) ou transferidos para AVSOLA a cada 8 semanas (referido como grupo de tratamento Remicade/AVSOLA). Os pacientes inicialmente randomizados com AVSOLA continuaram recebendo este tratamento a cada 8 semanas (referido como grupo de tratamento AVSOLA/AVSOLA). Os pacientes continuaram em tratamento até a semana 46. Uma única transição de Remicade para AVSOLA foi incorporada no desenho deste estudo a fim de verificar qualquer possível impacto desta transição na eficácia, segurança e imunogenicidade.
Resultado do Estudo
O desfecho primário de eficácia foi a diferença da resposta (DR) de 20% de melhora no critério de resposta (ACR20) do Colégio Americano de Reumatologia (ACR) analisados na semana 22 e na análise dos paramêtros de intenção de tratar (ITT) com imputação sem resposta para pacientes com falta de resposta ACR20 na semana 22. O desfechos secundários foram DR de ACR20 nas visitas agendadas diferente da semana 22; DR do ACR50 e ACR70, e alteração a partir do nível basal durante o estudo do Escore de Atividade da doença Disease Activity Score em 28 articulações - Proteína C reativa (DAS28-CRP). A equivalência clínica para o desfecho primário foi avaliado através da comparação do IC 90% para DR do ACR20 na semana 22 entre AVSOLA e Remicade com margem de equivalência de (-15%, 15%).
Na semana 22, 190 (68,1%) dos pacientes no grupo de tratamento de AVSOLA e 165 (59,1%) pacientes no grupo de tratamento Remicade atingiram o critério de resposta ACR20. A DR estimada do ACR20 entre AVSOLA e Remicade na semana 22 (Tabela 1) foi de 9,37% com IC 90% de (2,67%, 15,96%). O IC 90% foi ligeiramente excedido em seu limite superior pré-especificado da margem de equivalência (-15%, 15%). Análises adicionais ad-hoc do ACR20 na semana 22 foram conduzidas a fim de ajustar o impacto de um desbalanço aletório na linha de base demográfica e nas características da doença entre os 2 grupos de tratamento. Após o ajuste das características basais, a diferença observada foi reduzida e o IC 90% foi estreitada (7,18 [0,75, 13,62]) e desta forma permaneceu na margem de equivalência pré-especificada.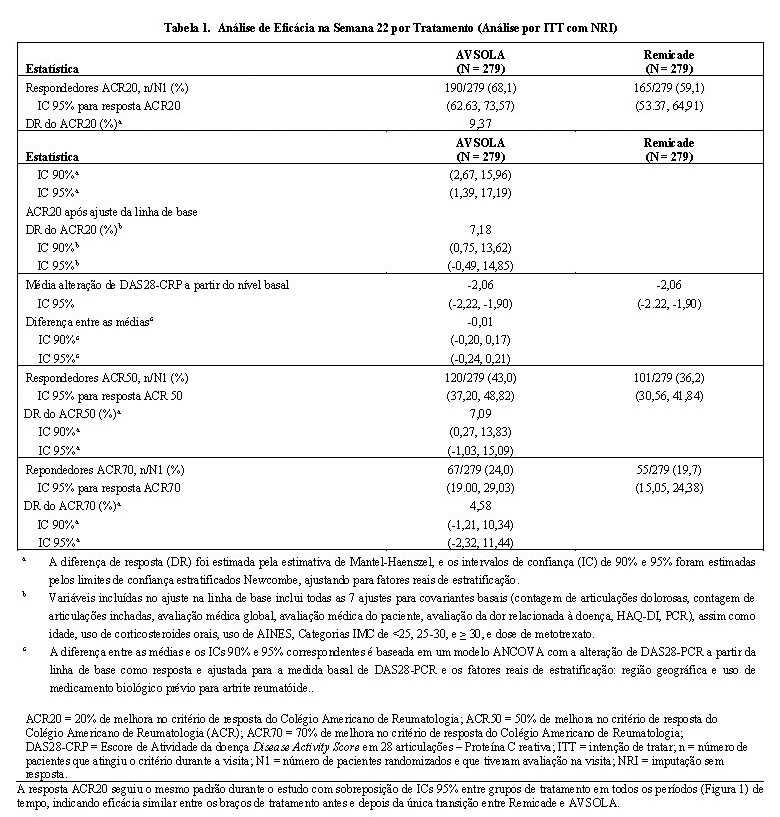
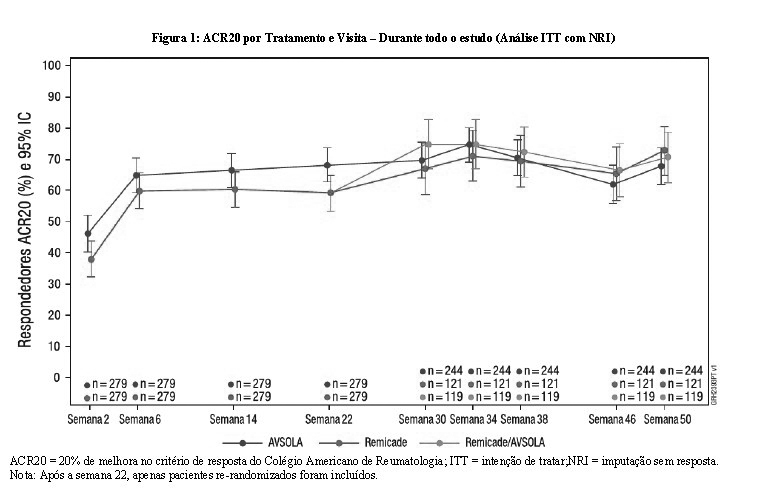
Durante todo o estudo, diferenças entre as médias nos desfechos secundários chave do DAS28-CRP foram mínimas em todos os períodos de tempo. Especificamente, na semana 22 (Tabela 3), a diferença entre grupos de tratamento de AVSOLA e Remicade na alteração média de DAS28-CRP a partir do nível basal foi de-0,01 com IC 90% (-0,20, 0,17) que demonstra a similaridade clínica.
Conforme mostrado na Figura 2, a alteração do DAS28-CRP a partir da linha de base por grupos de tratamento se sobrepôs ao longo de todo o estudo. Os resultados mostram que a eficácia foi mantida de forma consistente ao longo do estudo. A única transição de infliximabe para AVSOLA não afetou a eficácia.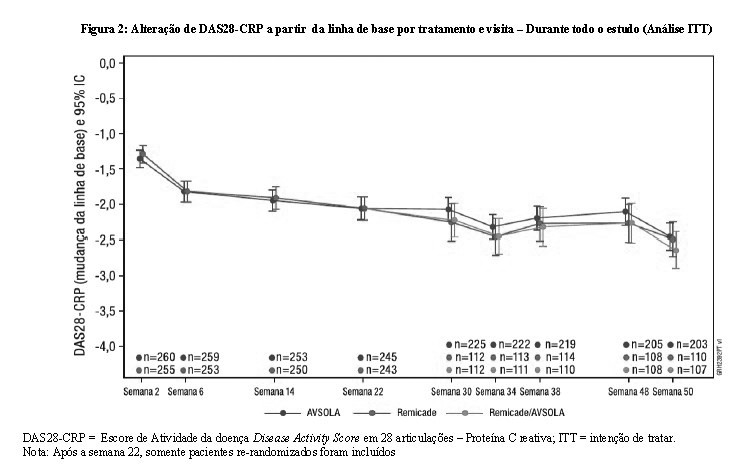
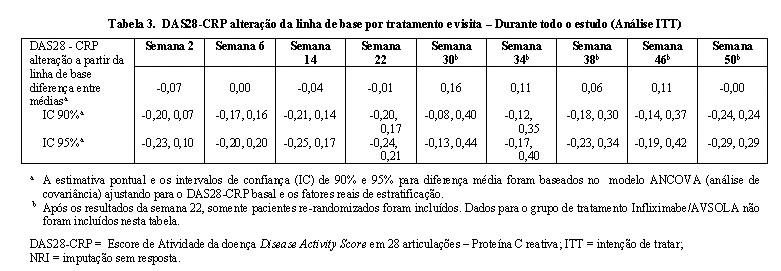
Os resultados estatísticos do desfecho primário na semana 22 confirmaram que AVSOLA é não inferior ao Remicade, entretanto a DR estimada do ACR20 entre AVSOLA e Remicade na semana 22 foi de 9,37% com IC 90% de (2,67%,15,96%), excedendo ligeiramente o limite superior da margem de equivalência (-15%, 15%). Após ajuste devido a um ligeiro desbalanço no nível basal das características, a diferença na resposta do ACR20 na semana 22 foi estreitada e contida na margem de equivalência pré-especificada. A totalidade dos dados de eficácia do ACR 20, ACR 50, ACR 70 e DAS28-CRP nos pontos de tempo suportam a conclusão da similaridade sem diferenças clínicas significativas de eficácia entre AVSOLA e Remicade. Uma única transição entre Remicade e AVSOLA não impactou a eficácia.
Imunogenicidade
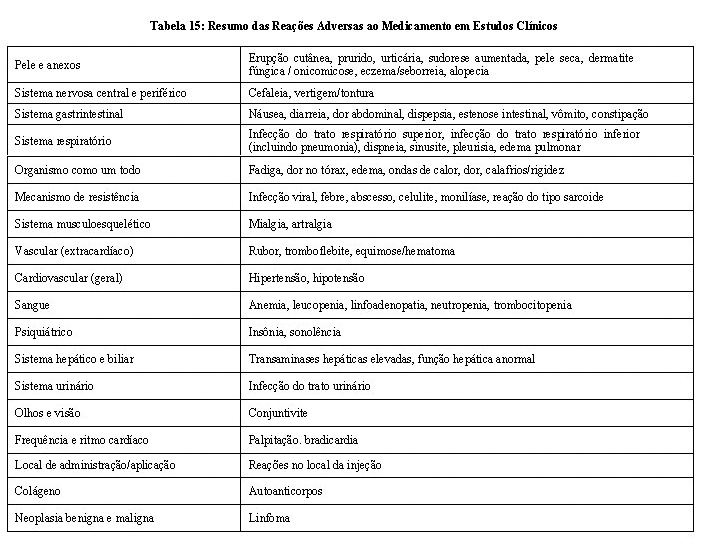
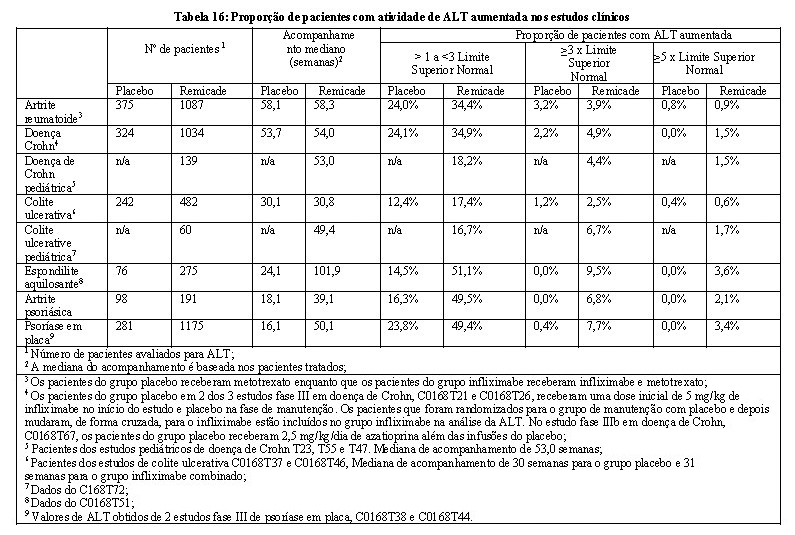
Assim como todas proteínas terapêuticas, há potencial imunogenicidade. Diferenças na metodologia do ensaio para medir a imunogenicidade impedem a comparação direta das taxas de imunogenicidade entre o AVSOLA e o Remicade ou outros agentes biológicos em diferentes estudos. No estudo 20140111, a atividade de anticorpos anti-medicamento (ADA) foi determinada utilizando imunoensaio de eletroquimioluminescência capaz de detectar anticorpos de pacientes tratados com AVSOLA e Remicade, e a atividade neutralizante dos ADA foi determinada utilizando um único ensaio de ligação ao alvo de AVSOLA capaz de determinar o anticorpo neutralizante.
No estudo 20140111 pacientes com artrite reumatoide de moderada a severa, a incidência de anticorpos para AVSOLA foi similar a Remicade. Pós nível basal (até a semana 22), 149 (57,1%) indivíduos no grupo de tratamento de AVSOLA e 160 (60,6%) indivíduos no grupo de tratamento de Remicade
testaram positivo para o desenvolvimento de ADAs ligantes; os resultados foram transitórios (ex: o resultado do ADA do indivíduo foi negativo para o último ponto de tempo testado no período do estudo) para 18 (6,9%) indivíduos no grupo de tratamento de AVSOLA, e 12 (4,5%) indivíduos no grupo de tratamento de Remicade. Pós nível basal (até a semana 22), 47 (18,0%) dos indivíduos no grupo de tratamento de AVSOLA e 55 (20,8%) dos indivíduos do grupo de tratamento de Remicade testaram positivo para o desenvolvimento de ADAs neutralizantes; os resultados foram transitórios (ex: o resultado do ADA do indivíduo foi negativo para o último ponto de tempo testado no período do estudo) para 5 (1,9%) dos indivíduos no grupo de tratamento de AVSOLA e 3 (1,1%) dos indivíduos no grupo de tratamento de Remicade.
Após a semana 22, um total de 66 (35,5%) indivíduos (29 [30,2%] no grupo de tratamento AVSOLA/AVSOLA, 19 [42,2%] no grupo de tratamento Remicade/Remicade, e 18 [40,0%] no grupo de tratamento Remicade/AVSOLA) testaram positivo para o desenvolvimento de ADAs. Os resultados foram transitórios (ex: o resultado do ADA do indivíduo foi negativo para o último ponto de tempo testado no período do estudo) para 8 indivíduos [8,3%] no grupo de tratamento AVSOLA/AVSOLA, 7 indivíduos [15,6%] no grupo de tratamento Remicade/Remicade, e 9 indivíduos [20,0%] no grupo de tratamento Remicade/AVSOLA.
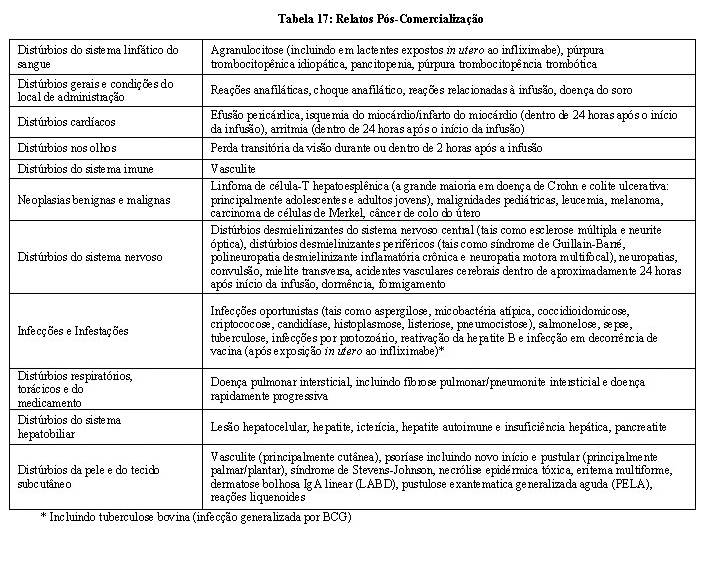
Resultados de ensaio de imunogenicidade são altamente dependentes da sensibilidade e especificidade do método de teste e podem ser influenciados por diversos fatores, incluindo manuseio de amostra, tempo de coleta de amostra, medicações concomitantes e doença subjacente. Por essas razões, a comparação da incidência de anticorpos ao AVSOLA com a incidência de anticorpos a outros produtos pode ser equivocada.
Dados de Eficácia para Remicade
Artrite reumatoide
A segurança e a eficácia de Remicade foram avaliadas em 2 estudos pivotais, multicêntricos, randomizados, duplo-cego: ATTRACT (Estudo AR I) e ASPIRE (Estudo AR II). Foi permitido uso concomitante de doses estáveis de ácido fólico, corticosteroides orais (≤ 10 mg/dia) e/ou medicamentos antiinflamatórios não esteroidais (AINEs).
O Estudo AR I foi um estudo controlado com placebo em 428 pacientes com artrite reumatoide ativa, apesar do tratamento com metotrexato (MTX). Os pacientes incluídos no estudo tinham uma idade mediana de 54 anos, duração mediana da doença de 8,4 anos, mediana de articulações dolorosas e edemaciadas de 20 e 31, respectivamente, e estavam sob tratamento com dose mediana de metotrexato de 15 mg por semana. Os pacientes receberam tanto placebo + metotrexato ou uma de 4 doses/esquemas de tratamento com Remicade + MTX:3 mg/kg ou 10 mg/kg de Remicade por infusão intravenosa nas semanas 0, 2 e 6, seguidas por infusões adicionais a cada 4 ou 8 semanas, em combinação com metotrexato.
O Estudo AR II foi um estudo controlado com placebo, de 3 braços ativos, em 1004 pacientes com artrite reumatoide ativa com duração mediana da doença de 3 anos ou menos, virgens de tratamento com metotrexato.
Os pacientes incluídos tinham idade mediana de 51 anos com mediana de duração da doença de 0,6 anos e mediana de contagem de articulações dolorosas e edemaciadas de 19 e 31, respectivamente. Mais de 80% dos pacientes tinham erosões articulares no nível basal. Durante a randomização, todos os pacientes receberam metotrexato (otimizado com 20 mg/semana, até a semana 8) e também placebo, 3 mg/kg ou 6 mg/kg de Remicade nas semanas 0, 2 e 6; e depois a cada 8 semanas.
Os dados do uso de Remicade sem uso concomitante com metotrexato são limitados.
- Resposta Clínica:
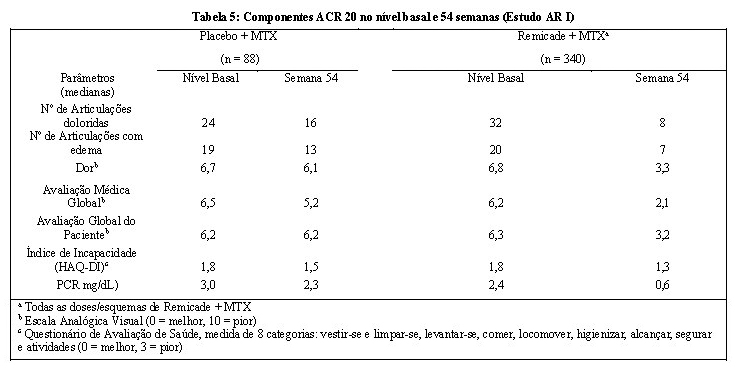
No Estudo AR I todas as doses/esquemas de Remicade + MTX resultaram em uma melhora nos sinais e sintomas, conforme medido pelo critério de resposta do Colégio Americano de Reumatologia (ACR 20), com uma maior porcentagem dos pacientes alcançando respostas ACR 20, 50 e 70 comparado com placebo + MTX (Tabela 4). Esta melhora foi observada na semana 2 e foi mantida até a semana 102. Foram observados maiores efeitos em cada componente do ACR 20 em todos os pacientes tratados com Remicade + MTX, comparado com placebo + MTX (Tabela 5). Mais pacientes tratados com Remicade alcançaram maior resposta clínica do que os pacientes tratados com placebo (Tabela 4).
No Estudo AR II, após 54 semanas de tratamento, ambas as doses de Remicade + MTX resultaram em maior resposta estatisticamente significativa nos sinais e sintomas, quando comparado com MTX em monoterapia, conforme medido pela proporção de pacientes que alcançaram respostas ACR 20, 50 e 70 (Tabela 4). Mais pacientes tratados com Remicade alcançaram uma resposta clínica melhor do que os pacientes tratados com placebo (Tabela 4).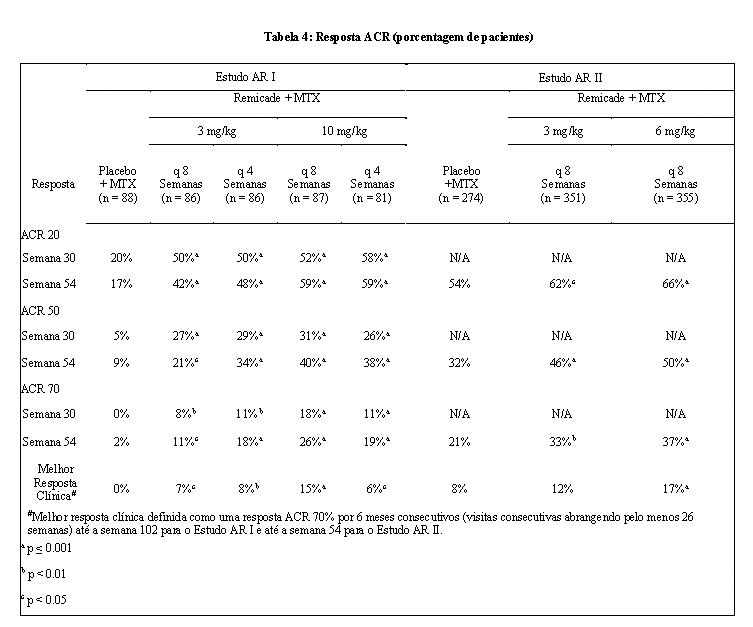
- Resposta Radiográfica
O dano estrutural em ambos mãos e pés foi avaliado radiograficamente na semana 54, pela alteração em relação ao nível basal na pontuação total van der Heidje modificado por Sharp (vdH-S), uma pontuação composta para dano estrutural que mede o número e tamanho de articulações com erosão, bem como o grau de estreitamento no espaço articular nas mãos/punhos e pés.
No Estudo AR I, aproximadamente 80% dos pacientes tiveram dados pareados de Raio-X combinados nas 54 semanas e aproximadamente 70% nas 102 semanas. A inibição na progressão do dano estrutural foi observada na semana 54 (Tabela 6) e mantida pelas 102 semanas.
No Estudo AR II, > 90% dos pacientes tinham ao menos 2 avaliações de Raio-X. A inibição na progressão do dano estrutural foi observada na semana 30 e na semana 54 (Tabela 6) nos grupos de Remicade + MTX, comparado com MTX em monoterapia. Os pacientes tratados com Remicade + MTX demonstraram menor progressão no dano estrutural comparado com MTX em monoterapia, quando os níveis basais de proteínas de fase aguda (Proteína C reativa (PCR)) e Velocidade de hemossedimentação (VHS) estavam normais ou elevados: os pacientes com níveis basais elevados de proteínas de fase aguda tratados com MTX em monoterapia demonstraram uma progressão média na pontuação total vdH-S de 4,2 unidades comparado com pacientes tratados com Remicade + MTX, que apresentaram 0,5 unidade de progressão. Pacientes com níveis basais normais de PCR tratados com MTX em monoterapia demonstraram progressão média na pontuação total vdH-S de 1,80 unidades, comparado com Remicade + MTX que apresentaram 0,2 unidades de progressão. Dos pacientes recebendo Remicade + MTX, 59% não tiveram progressão (pontuação total vdH-S ≤ 0 unidades) no dano estrutural comparado a 45% dos pacientes recebendo MTX em monoterapia. Em um subgrupo de pacientes que iniciaram o estudo sem erosões articulares, os pacientes tratados com Remicade + MTX mantiveram o estado de "sem erosão articular" no primeiro ano em uma maior proporção de pacientes, do que no grupo de MTX em monoterapia, 79% (77/98) vs. 58% (23/40), respectivamente (p < 0,01). Menos pacientes nos grupos Remicade + MTX (47%) desenvolveram erosões nas articulações não comprometidas, comparado ao MTX em monoterapia (59%).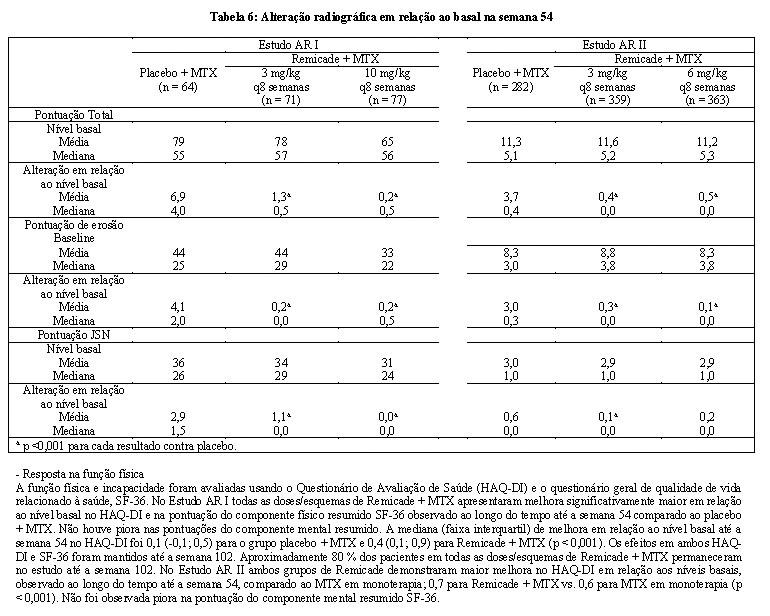
B>Espondilite anquilosante
A segurança e a eficácia de Remicade foram avaliadas em um estudo controlado com placebo, duplo-cego, multicêntrico, randomizado em 279 pacientes com espondilite anquilosante ativa. Os pacientes tinham entre 18 e 74 anos de idade e tinham espondilite anquilosante, conforme definido pelo critério modificado New York para Espondilite Anquilosante. Os pacientes tinham que ter doença ativa, conforme evidenciado pela pontuação no Índice de Atividade da Doença de Espondilite Anquilosante de Bath (BASDAI) > 4 (faixa possível de 0-10) e dor espinhal > 4 [Escala Visual Analógica (VAS) de 0-10]. Os pacientes com anquilose completa da coluna foram excluídos do estudo e foi proibido o uso de drogas modificadoras da doença (DMARDs) e corticosteroides de uso sistêmico. As doses de Remicade 5 mg/kg ou placebo foram administradas por via intravenosa nas semanas 0, 2, 6, 12 e 18. Em 24 semanas, foi observada melhora nos sinais e sintomas da espondilite anquilosante em 60% dos pacientes do grupo tratado com Remicade vs. 18% dos pacientes do grupo placebo (p < 0,001), conforme medido pela proporção de pacientes alcançando uma melhora de 20% no critério de resposta ASAS (ASAS 20). A melhora foi observada na semana 2 e mantida até a semana 24 (Figura 3 e Tabela 7).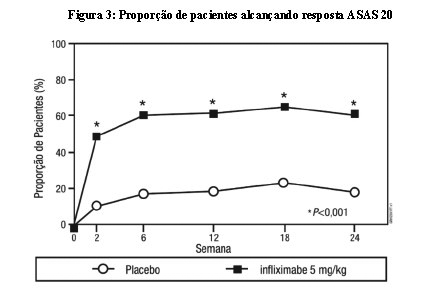
Em 24 semanas, a proporção de pacientes alcançando uma melhora nos sinais e sintomas de espondilite anquilosante de 50 % e 70%, conforme medido pelo critério de resposta ASAS (ASAS 50 e ASAS 70, respectivamente), foi de 44% e 28%, respectivamente, para os pacientes recebendo Remicade, comparado com 9% e 4%, respectivamente, para pacientes recebendo placebo (p < 0,001, Remicade vs. placebo). Um menor nível de atividade da doença [definido como um valor < 20 (na escala de 0-100 mm) em cada um dos 4 parâmetros de resposta ASAS] foi alcançado em 22% dos pacientes tratados com Remicade vs. 1% nos pacientes tratados com placebo (p < 0,001).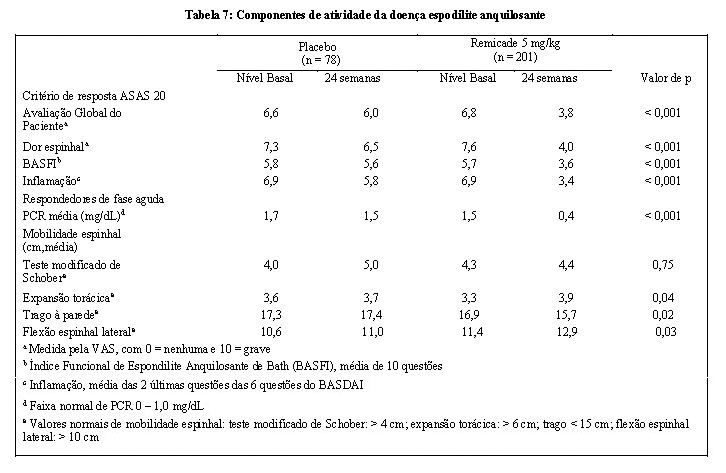
A melhora mediana em relação ao nível basal na pontuação do componente físico resumido no questionário geral de qualidade de vida relacionado à saúde, SF-36 na semana 24 foi de 10,2 para o grupo de tratamento com Remicade vs. 0,8 para o grupo placebo (p < 0,001). Não houve alteração na pontuação do componente mental resumido tanto para o grupo de Remicade como para o grupo placebo. Os resultados deste estudo foram semelhantes àqueles observados em um estudo multicêntrico, duplo-cego, controlado por placebo em 70 pacientes com espondilite anquilosante.
Artrite psoriática
A segurança e a eficácia de Remicade foram avaliadas em um estudo multicêntrico, duplo-cego, controlado com placebo em 200 pacientes adultos com artrite psoriásica ativa, apesar do tratamento com DMARDs ou AINE (≥ 5 articulações edemaciadas e ≥ 5 articulações dolorosas), com 1 ou mais dos seguintes subtipos: artrite acometendo articulações interfalangianas distais (DIP) (n= 49), artrite mutilante (n = 3), artrite periférica assimétrica (n = 40), artrite poliarticular (n = 100) e espondilite com artrite periférica (n = 8). Os pacientes também tinham psoríase em placa com uma lesão alvo de qualificação de diâmetro ≥ 2 cm. Quarenta e seis por cento dos pacientes continuaram em doses estáveis de metotrexato (≤ 25 mg/semana). Durante a fase duplo cego de 24 semanas, os pacientes receberam tanto 5 mg/kg de Remicade ou placebo nas semanas 0, 2, 6, 14 e 22 (100 pacientes em cada grupo). Na semana 16, os pacientes com < 10% de melhora em relação aos níveis basais, em ambas contagens de articulações edemaciadas e articulações dolorosas, passaram a receber indução com Remicade (escape precoce). Na semana 24, todos os pacientes do grupo placebo passaram a receber indução com Remicade. A administração continuou para todos os pacientes até a semana 46.
- Resposta Clínica
O tratamento com Remicade resultou em melhora nos sinais e sintomas, conforme avaliado pelo critério ACR, com 58% dos pacientes do grupo tratado com Remicade alcançando ACR 20 na semana 14, comparado com 11% dos pacientes tratados com placebo (p < 0,001). A resposta foi semelhante, independente do uso concomitante de metotrexato. Foi observada melhora no início da semana 2. Em 6 meses, as respostas ACR 20/50/70 foram alcançadas por 54%, 41% e 27%, respectivamente, dos pacientes recebendo Remicade comparado com 16%, 4% e 2%, respectivamente, dos pacientes recebendo placebo. Respostas semelhantes foram observadas em pacientes com cada subtipo de artrite psoriásica, entretanto, poucos pacientes com os subtipos artrite mutilante e espondilite com artrite periférica foram incluídos. Quando comparado ao placebo, o tratamento com Remicade resultou em melhora nos componentes do critério de resposta ACR, assim como em dactilites e entesopatia (Tabela 8). A resposta clínica foi mantida ao longo das 54 semanas. Respostas semelhantes foram observadas na fase inicial do estudo randomizado, controlado com placebo em 104 pacientes com artrite psoriásica e as respostas foram mantidas ao longo de 98 semanas na fase de extensão aberta.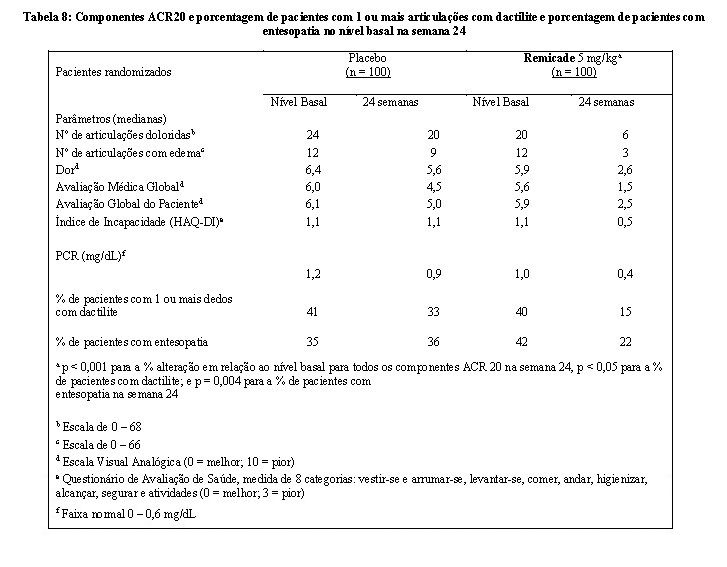
A melhora no Índice de Gravidade e Área de Psoríase (PASI) em pacientes com artrite psoriásica com área de superfície corporal (BSA) ≥ 3 (placebo n = 87, Remicade n = 83) foi alcançada na semana 14, independente do uso concomitante de metotrexato, com 64% dos pacientes tratados com Remicade alcançando 75% de melhora em relação aos níveis basais vs. 2% dos pacientes tratados com placebo. Uma melhora foi observada em alguns pacientes no início da semana 2. Em 6 meses, as respostas PASI 75 e PASI 90 foram alcançadas por 60% e 39%, respectivamente, para os pacientes tratados com Remicade comparado a 1% e 0%, respectivamente, dos pacientes recebendo placebo. A resposta PASI foi em geral mantida até a semana 54.
- Resposta radiográfica
O dano estrutural em ambos mãos e pés foi avaliado radiograficamente pela alteração em relação aos níveis basais na pontuação total modificada vdH-S incluindo articulações DIP. A pontuação total modificada vdH-S é uma pontuação composta de dano estrutural que mede o número e extensão das erosões nas articulações e o grau de estreitamento do espaço articular (JSN) nas mãos e pés. Na semana 24, os pacientes tratados com Remicade tiveram menos progressão radiográfica do que os pacientes tratados com placebo (alteração média de -0,70 vs. 0,82; p < 0,001). Os pacientes tratados com Remicade também tiveram menos progressão em suas pontuações de erosão (-0,56 vs. 0,51) e pontuações JSN (-0,14 vs. 0,31). Os pacientes no grupo de Remicade apresentaram inibição contínua no dano estrutural na semana 54. A maioria dos pacientes demonstrou pequena ou nenhuma alteração na pontuação vdH-S durante os 12 meses de estudo (alteração mediana de 0 em ambos pacientes que inicialmente receberam Remicade ou placebo). Mais pacientes do grupo placebo (12%) tiveram progressão aparente imediata, comparada ao grupo de Remicade (3%).
- Função Física
O status da função física foi avaliado usando o Índice de Incapacidade HAQ (HAQ-DI) e o questionário de saúde SF-36. Os pacientes tratados com Remicade tiveram melhora significativa na função física, conforme avaliado pelo HAQ-DI (porcentagem mediana de melhora na pontuação HAQ-DI em relação aos níveis basais na semana 14 e 24 de 43% dos pacientes tratados com Remicade vs. 0% dos pacientes tratados com placebo). Durante a fase duplo-cega do estudo, controlada com placebo (24 semanas), 54% dos pacientes tratados com Remicade alcançaram melhora clinicamente significativa no HAQ-DI (diminuição de ≥ 0,3 unidades) comparado com 22% dos pacientes tratados com placebo. Os pacientes tratados com Remicade também demonstraram maior melhora nas pontuações dos componentes mental e físico resumidos SF-36, do que os pacientes tratados com placebo. As respostas foram mantidas por até 2 anos na fase de extensão aberta do estudo.
Psoríase em placa
A segurança e a eficácia de Remicade foram avaliadas em 3 estudos randomizados, duplo-cegos, controlados com placebo em pacientes com 18 anos ou mais, com psoríase em placa crônica estável acometendo ≥ 10% BSA, uma pontuação PASI mínima de 12 e que eram candidatos para terapia sistêmica ou fototerapia. Os pacientes com psoríase gutata, pustular ou eritrodérmica foram excluídos destes estudos. Nenhuma terapia antipsoriásica foi permitida durante o estudo, com exceção de corticosteroides tópicos de baixa potência na face e virilha após a semana 10 do início do estudo.
O Estudo I (EXPRESS) avaliou 378 pacientes que receberam placebo ou Remicade na dose de 5 mg/kg nas semanas 0, 2 e 6 (terapia de indução), seguida por terapia de manutenção a cada 8 semanas. Na semana 24, o grupo placebo passou a receber terapia de indução com Remicade (5 mg/kg), seguida pela terapia de manutenção a cada 8 semanas. Os pacientes originalmente randomizados para Remicade continuaram a receber Remicade 5 mg/kg a cada 8 semanas até a semana 46. Entre todos os grupos de tratamento, a pontuação PASI mediana no nível basal foi de 21 e a pontuação da Avaliação Médica Global Estática (sPGA) no nível basal variou de moderada (52% dos pacientes) para intensa (36%), para grave (2%). Adicionalmente, 75% dos pacientes tiveram um BSA > 20%. Setenta e um por cento dos pacientes previamente receberam tratamento sistêmico e 82% dos pacientes receberam fototerapia. O Estudo II (EXPRESS II) avaliou 835 pacientes que receberam placebo ou Remicade nas doses de 3 mg/kg ou 5 mg/kg, nas semanas 0, 2 e 6 (terapia de indução). Na semana 14, dentro de cada grupo de dose de Remicade, os pacientes foram randomizados ou para os esquemas de tratamento (a cada 8 semanas), ou tratamento de manutenção conforme necessário (PRN) até a semana 46. Na semana 16, o grupo placebo passou a receber terapia de indução de Remicade (5 mg/kg), seguido de terapia de manutenção a cada 8 semanas. Entre todos os grupos de tratamento, a mediana na pontuação PASI basal foi 18 e 63% dos pacientes tiveram uma BSA > 20%. Cinquenta e cinco por cento dos pacientes previamente receberam terapia sistêmica e 64% receberam uma fototerapia.
Um Estudo III (SPIRIT) avaliou 249 pacientes que tinham recebido previamente ou tratamento com psoraleno + ultravioleta A (PUVA), ou outra terapia sistêmica para psoríase. Estes pacientes foram randomizados para receber placebo ou Remicade nas doses 3 mg/kg ou 5 mg/kg, nas semanas 0, 2 e 6. Na semana 26, os pacientes com pontuação sPGA moderada ou pior (maior que ou igual a 3 em uma escala de 0 a 5) receberam uma dose adicional do tratamento randomizado. Entre todos os grupos de tratamento, a pontuação PASI mediana no nível basal foi de 19 e a pontuação sPGA no nível basal variou de moderada (62% dos pacientes) para intensa (22%), para grave (3%). Adicionalmente, 75% dos pacientes tiveram um BSA > 20%.
Dos pacientes incluídos, 114 (46%) receberam dose adicional na semana 26. Nos Estudos I, II e III, o desfecho primário foi a proporção de pacientes que alcançaram uma redução na pontuação PASI de ao menos 75% (PASI 75) em relação ao nível basal, na semana 10. No Estudo I e Estudo III, outro fator avaliado incluiu a proporção de pacientes que atingiram uma pontuação de sPGA "clareamento completo" ou "doença mínima". A escala sPGA é uma escala de 6 categorias, variando de "5 = doença grave" a "0 = clareamento completo", que indica a avaliação global do médico da gravidade da psoríase, com foco na infiltração, eritema e descamação. O sucesso do tratamento, definido como "clareamento completo" ou "doença mínima", consistiu de nenhuma ou mínima elevação da placa, com eritema de coloração até vermelho claro e nenhuma ou mínima descamação fina de < 5% da superfície da placa.
O Estudo II também avaliou a proporção de pacientes que alcançou uma pontuação de "clareamento completo" ou "excelente" pela Avaliação Global Médica relativa (rPGA). O rPGA é uma escala de 6 categorias que varia de "6 = piora" a "1 = clareamento completo" que foi avaliada em relação ao nível basal. As lesões globais foram classificadas conforme a porcentagem de acometimento corporal, bem como a infiltração global, descamação e eritema. O sucesso do tratamento, definido como "clareamento completo" ou "excelente", consistiu em alguma mudança de coloração residual rosada ou pigmentação, para melhora acentuada (textura da pele próxima do normal. Algum eritema pode estar presente). Os resultados destes estudos estão apresentados na Tabela 9.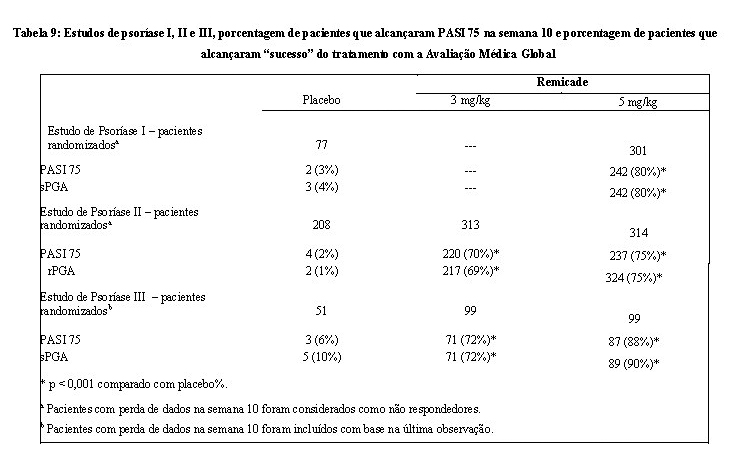
No Estudo I, no subgrupo de pacientes com maior extensão da psoríase que tinham recebido fototerapia prévia, 85% dos pacientes do regime de 5 mg/kg de Remicade alcançaram PASI 75 na semana 10, comparado a 4% dos pacientes do grupo placebo.
No Estudo II, no subgrupo de pacientes com maior extensão da psoríase que tinham recebido fototerapia prévia, 72% e 77% dos pacientes no regime de 3 mg/kg de Remicade e 5 mg/kg de Remicade alcançaram PASI 75 na semana 10, respectivamente, comparado a 1% no grupo placebo. No Estudo II, entre os pacientes com maior extensão da psoríase que falharam ou eram intolerantes à fototerapia, 70% e 78% dos pacientes no regime de 3 mg/kg de Remicade e 5 mg/kg de Remicade alcançaram PASI 75 na semana 10, respectivamente, comparado a 2% no grupo placebo.
A manutenção da resposta foi estudada em subgrupos de 292 e 297 pacientes tratados com Remicade nos grupos de 5 mg/kg e 3 mg/kg, respectivamente, no Estudo II. No início da semana 14, com base nas respostas estratificadas por PASI na semana 10 e por centro de pesquisa, os pacientes dos grupos ativos foram randomizados novamente para ambos regimes de dose ou para o tratamento de manutenção necessário (PRN).
Os grupos que receberam a dose de manutenção a cada 8 semanas parecem ter tido maior porcentagem de do PASI 75 sustentada até a semana 50, quando comparado com aqueles pacientes que receberam doses conforme necessário ou PRN, e a melhor resposta foi mantida para a dose de 5 mg/kg a cada 8 semanas. Estes resultados são apresentados na Figura 4. Na semana 46, quando as concentrações séricas de Remicade estavam no vale no grupo de dose a cada 8 semanas, 54% dos pacientes do grupo de 5 mg/kg alcançaram PASI 75 comparado com 36% no grupo de 3 mg/kg. A menor porcentagem de respondedores PASI 75 no grupo de dose 3 mg/kg a cada 8 semanas, comparado ao grupo de 5 mg/kg foi associada a menor porcentagem de pacientes com níveis séricos (vale) de infliximabe detectável. Isto pode estar relacionado em parte com as maiores taxas de anticorpos. Além disso, no subgrupo de pacientes que alcançaram uma resposta na semana 10, a manutenção da resposta parece ser maior nos pacientes que receberam Remicade a cada 8 semanas na dose de 5 mg/kg. Independente se as doses de manutenção são PRN ou a cada 8 semanas, há uma diminuição na resposta da subpopulação de pacientes em cada grupo ao longo do tempo. Os resultados do Estudo I até a semana 50 nos grupos de regime de dose de manutenção de 5 mg/kg a cada 8 semanas, foram semelhantes aos resultados obtidos no Estudo II.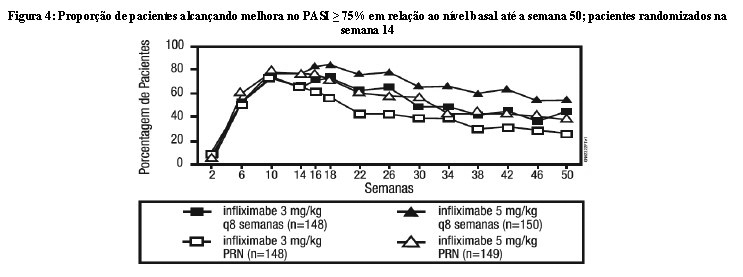
A eficácia e segurança do tratamento de Remicade além de 50 semanas não foram avaliadas em pacientes com psoríase em placa.
Doença de Crohn ativa
A segurança e eficácia de doses únicas e múltiplas de Remicade foram avaliadas em 2 estudos clínicos randomizados, duplo-cegos, controlados com placebo, em 653 pacientes com doença de Crohn gravemente ativa [Índice de Atividade da Doença de Crohn (CDAI) ≥ 220 e ≤ 400] com resposta inadequada para terapias convencionais prévias. Foi permitido o uso concomitante de doses estáveis de aminossalicilatos, corticosteroides e/ou agentes imunomoduladores e 92% dos pacientes continuaram a receber ao menos uma dose destas medicações.
No estudo de dose única com 108 pacientes, 16% (4/25) dos pacientes do grupo placebo alcançaram uma resposta clínica (diminuição no CDAI ≥ 70 pontos) na semana 4 vs. 81% (22/27) dos pacientes recebendo 5 mg/kg de Remicade (p < 0,001, Teste Exato de Fisher, 2 x 2). Adicionalmente, 4% (1/25) dos pacientes do grupo placebo e 48% (13/27) dos pacientes recebendo 5 mg/kg de Remicade alcançaram remissão clínica (CDAI < 150) na semana 4.
Em um estudo de multidose [ACCENT 1 (Estudo de Crohn I)], 545 pacientes receberam 5 mg/kg na semana 0 e então foram randomizados para um de três grupos de tratamento. O grupo de manutenção com placebo recebeu placebo nas semanas 2 e 6 e depois, a cada 8 semanas. O grupo de manutenção com 5 mg/kg recebeu 5 mg/kg nas semanas 2 e 6 e depois, a cada 8 semanas. O grupo de manutenção com 10 mg/kg recebeu 5 mg/kg nas semanas 2 e 6 e depois, 10 mg/kg a cada 8 semanas. Os pacientes em resposta na semana 2 foram randomizados e analisados separadamente daqueles que não responderam nesta semana. Após a semana 6, a retirada gradual de corticosteroides foi permitida.
Na semana 2, 57% (311/545) dos pacientes estavam em resposta clínica. Na semana 30, uma proporção significativamente maior destes pacientes nos grupos de manutenção com 5 mg/kg e 10 mg/kg alcançou remissão clínica, comparado aos pacientes no grupo de manutenção com placebo (Tabela 10).
Adicionalmente, uma proporção significativamente maior de pacientes nos grupos de manutenção com Remicade 5 mg/kg e 10 mg/kg estavam em remissão clínica e aptos para interromper o uso de corticosteroides, quando comparados aos pacientes do grupo de manutenção com placebo, na semana 54 (Tabela 10).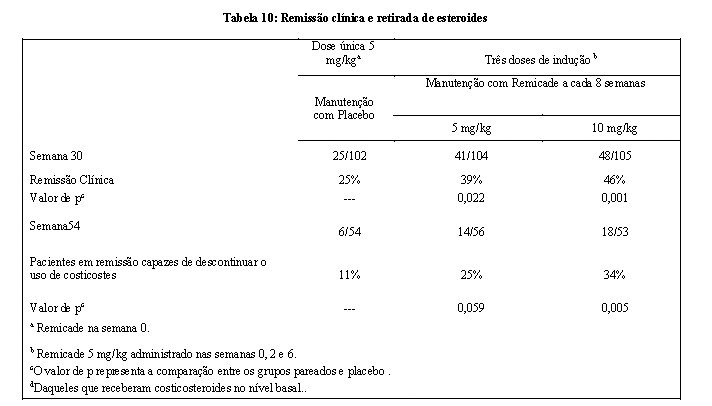
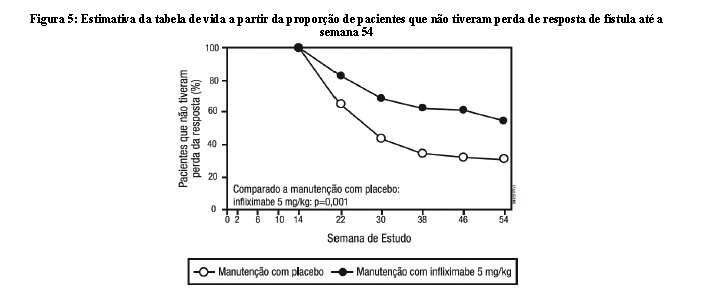
Os pacientes nos grupos de manutenção com Remicade (5 mg/kg e 10 mg/kg) tiveram um maior tempo para perder resposta do que os pacientes que estavam no grupo de manutenção com placebo (Figura 5). Na semana 30 e 54, uma melhora significativa em relação ao nível basal foi observada entre os grupos tratados com Remicade 5 mg/kg e 10 mg/kg, comparado com o grupo tratado com placebo, no questionário específico para doença inflamatória intestinal (IBQD), particularmente nos componentes sistêmicos e intestinais e na pontuação dos componentes físicos resumidos do questionário geral de qualidade de vida relacionado à saúde, SF-36.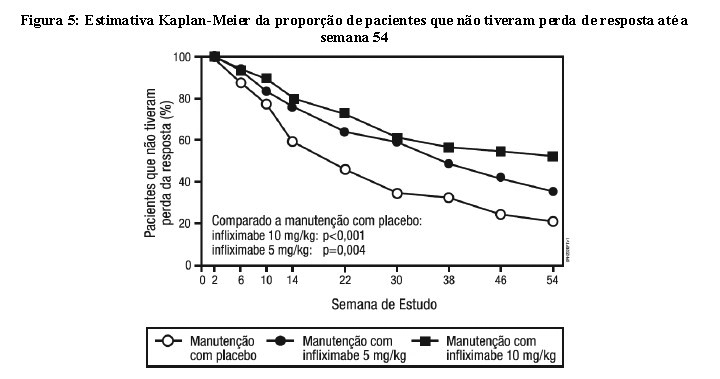
Em um subgrupo de 78 pacientes que tinham ulceração da mucosa no nível basal e que participaram de um subestudo de endoscopia, 13 de 43 pacientes do grupo de manutenção com Remicade tinham evidência de cicatrização da mucosa, quando comparado com 1 de 28 pacientes tratados com placebo, na semana 10. Dos pacientes tratados com Remicade que apresentavam cicatrização da mucosa na semana 10, 9 de 12 pacientes apresentaram cicatrização da mucosa na semana 54.
Os pacientes que alcançaram uma resposta e subsequentemente perderam, foram elegíveis ao tratamento com Remicade de forma episódica em uma dose 5 mg/kg maior do que a dose para a qual foram randomizados. A maioria destes pacientes respondeu a uma maior dose. Entre os pacientes que não tinham respondido na semana 2, dos que receberam doses de manutenção de Remicade, 59% (92/157) responderam até a semana 14 comparado a 51% (39/77) do grupo de manutenção com placebo. Entre os pacientes que não apresentaram resposta até a semana 14, a administração de tratamento adicional não resultou em aumento significativo de respostas.
Doença de Crohn fistulizante
A segurança e a eficácia de Remicade em pacientes com doença de Crohn fistulizante com fístula(s) que tinham ao menos 3 meses de duração, foram avaliadas em 2 estudos randomizados, duplo-cegos, controlados com placebo. Foi permitido o uso concomitante de doses estáveis de corticosteroides, 5-aminossalicilatos, antibióticos, metotrexato (MTX), 6-mercaptopurina (6-MP) e/ou azatioprina (AZA). No primeiro estudo, 94 pacientes receberam 3 doses de placebo ou Remicade nas semanas 0, 2 e 6. A resposta da fístula (≥ 50% da redução no número de fístulas de drenagem enterocutâneas por compressão leve, em ao menos 2 visitas consecutivas sem aumento de medicação ou cirurgia para doença de Crohn) foi observada em 68% (21/31) dos pacientes do grupo tratado com Remicade 5 mg/kg (p = 0,002) e 56% (18/32) dos pacientes tratados com Remicade 10 mg/kg (p=0,021) vs. 26% (8/31) dos pacientes do braço do placebo. A mediana para o tempo do início da resposta e a mediana da duração da resposta nos pacientes tratados com Remicade foi de 2 e 12 semanas, respectivamente. O fechamento de todas as fístulas foi alcançado em 52% dos pacientes tratados com Remicade comparado com 13% dos pacientes do grupo placebo (p < 0,001). No segundo estudo [ACCENT II (Estudo de Crohn II)], os pacientes que foram incluídos tiveram que ter ao menos 1 fístula de drenagem enterocutânea (perianal e abdominal). Todos os pacientes receberam Remicade nas semanas 0, 2 e 6. Os pacientes foram randomizados para manutenção com placebo ou Remicade 5 mg/kg na semana 14. Os pacientes receberam as doses de manutenção na semana 14 e depois a cada 8 semanas, até a semana 46. Os pacientes que tinham resposta da fístula (resposta da fístula foi definida da mesma forma que no primeiro estudo) nas semanas 10 e 14 foram randomizados separadamente daqueles que não tinham resposta. O desfecho primário foi o tempo para a perda da resposta a partir da randomização entre os pacientes que tinham resposta da fístula. Entre os pacientes randomizados (273 dos 296 pacientes inicialmente incluídos), 87% tinham fístulas perianais e 14% tinham fístulas abdominais. Oito por cento também tinham fístulas retovaginais. Mais de 90% dos pacientes tinham recebido tratamento prévio com imunossupressores ou antibióticos. Na semana 14, 65% (177/273) dos pacientes tinham respostas da fístula. Os pacientes randomizados para o tratamento de manutenção com Remicade tinham maior tempo para a perda da resposta da fístula comparado com o grupo de manutenção com placebo (Figura 5). Na semana 54, 38% (33/87) dos pacientes tratados com Remicade não tinham drenagem de fístulas, comparado com 22% (20/90) dos pacientes tratados com placebo (p = 0,