APRETUDE
GLAXOSMITHKLINE
Comprimidos
cabotegravir
Profilaxia pré-exposição ao HIV.
Apresentações.
Apretude® é apresentado na forma de comprimidos revestidos contendo 30 mg de cabotegravir em frasco com 30 comprimidos.
USO ORAL
USO ADULTO E PEDIÁTRICO (ACIMA DE 12 ANOS COM PESO MÍNIMO DE 35 KG)
Composição.
Cada comprimido revestido de Apretude® contém: cabotegravir30 mg (equivalentes a 31,62 mg de cabotegravir sódico), excipientes* q.s.p. 1 comprimido revestido
*lactose monoidratada, celulose microcristalina, hipromelose, amidoglicolato de sódio, estearato de magnésio, água purificada e Aquarius BP18237 Branco ou Opadry OY-S-28876 Branco (hipromelose, dióxido de titânio e macrogol).
Informações técnicas.
1. INDICAÇÕES
Apretude® comprimidos revestidos é indicado como medicamento preventivo, como parte de estratégia de prevenção combinada ao vírus da imunodeficiência humana (HIV), na profilaxia pré-exposição (PrEP) para reduzir o risco de HIV-1 adquirido sexualmente em adultos e adolescentes acima de 12 anos pesando pelo menos 35 kg e com risco aumentado de adquirir a infecção (ver Posologia e Modo de Usar, Advertências e Precauções).
Para entendimento de contextos de risco aumentado de aquisição do HIV-1, devem ser considerados entre outros, mas não restrito aos seguintes aspectos:
a.repetição de práticas sexuais anais ou vaginais com penetração sem o uso de preservativo;
b.frequência de relações sexuais com parcerias eventuais;
c.quantidade e diversidade de parcerias sexuais;
d.histórico de episódios de DST (doenças sexualmente transmissíveis)
e. busca repetida por profilaxia pós-exposição (PEP);
f. contextos de relações sexuais em troca de dinheiro, objetos de valor, drogas, moradia, entre outros serviços;
g. Chemsex: prática sexual sob a influência de drogas psicoativas (metanfetaminas, gama-hidroxibutirato - GHB, midomafetamina - MDMA, cocaína, poppers, entre outras) com a finalidade de melhorar ou facilitar as experiências sexuais.
Os comprimidos de Apretude® podem ser usados como:
-introdução oral para avaliar a tolerabilidade de cabotegravir antes da administração de cabotegravir injetável de ação prolongada (LA);
-terapia oral em indivíduos que irão perder a dose planejada de cabotegravir injetável.
2. RESULTADOS DE EFICÁCIA
Eficácia e segurança clínica
A eficácia de cabotegravir para profilaxia-pré exposição (PrEP) foi avaliada em dois estudos clínicos controlados, randomizados (1:1), duplocegos, multicêntricos e com dois braços. A eficácia de cabotegravir foi comparada com fumarato de tenofovir desoproxila (TDF)/entricitabina (FTC) oral diário. Os participantes randomizados para receber cabotegravir iniciaram a administração de introdução oral com um comprimido de 30 mg de cabotegravir e um placebo por dia, por até 5 semanas, seguido por injeção intramuscular (IM) de cabotegravir (injeção única de 600 mg [3 mL]), nos meses 1, 2 e a cada 2 meses depois disso e um comprimido de placebo diário. Participantes randomizados para receber TDF/FTC iniciaram com TDF 300 mg/FTC 200 mg oral e placebo por até 5 semanas, seguido por TDF 300 mg/FTC 200 mg oral diário e placebo injetável (IM) (3 mL, emulsão lipídica injetável a 20% nos meses 1, 2 e a cada 2 meses depois disso).
Estudo HPTN 083
No estudo HPTN 083, de não inferioridade, 4.566 homens cisgênero e mulheres transgênero que fazem sexo com homens foram randomizados
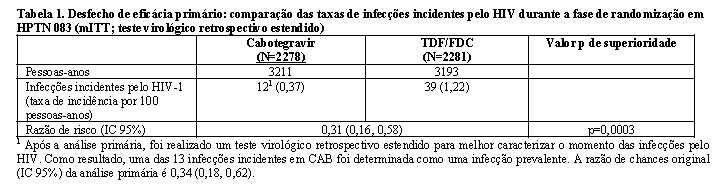
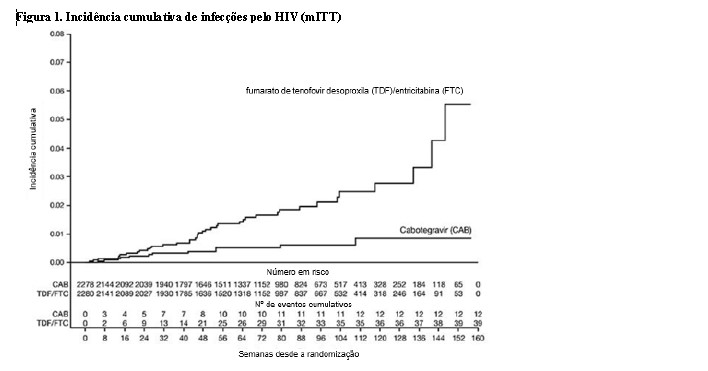
1:1 e receberam cabotegravir (n = 2281) ou TDF/FTC (n = 2285) como medicação do estudo cego até a Semana 153. Na visita basal, a mediana da idade dos participantes era de 26 anos, 12% eram mulheres transgênero, 72% eram não brancos (sendo 27% índios Americanos ou nativos do Alasca, 25% negros, 18% Asiáticos e 2% mistura de raças) e 67% tinham < 30 anos de idade. O desfecho primário foi a taxa de infecções incidentes pelo HIV entre os participantes randomizados para cabotegravir oral e cabotegravir injetável em comparação com TDF/FTC oral. A análise primária demonstrou a superioridade de cabotegravir em comparação com TDF/FTC (corrigido para interrupção prematura) com uma redução de 66% no risco de adquirir a infecção incidente pelo HIV, razão de chances (IC 95%) 0,34 (0,18, 0,62); testes adicionais revelaram que uma das infecções em quem utilizava cabotegravir era prevalente, resultando em uma redução de 69% no risco de infecção incidente em relação ao TDF/FTC (consulte a Tabela 1).
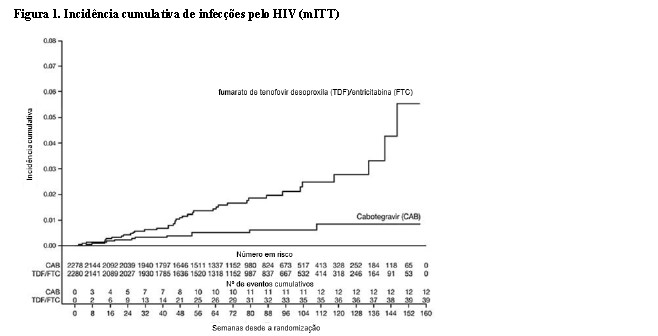
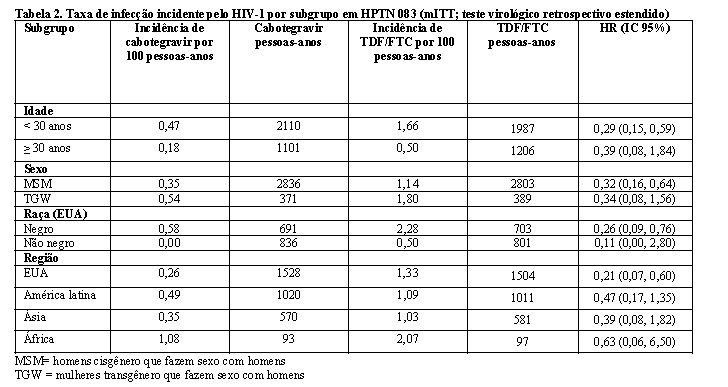
Os achados de todas as análises de subgrupos foram consistentes com o efeito protetor global, com uma taxa inferior de infecções incidentes pelo HIV-1 observada para participantes randomizados para o grupo cabotegravir em comparação com participantes randomizados para o grupo TDF/FTC (consulte a Tabela 2).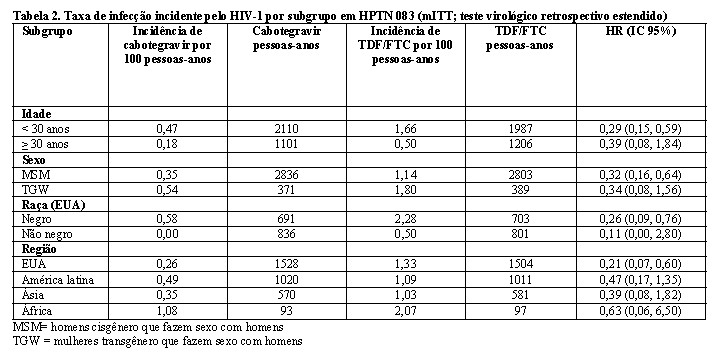
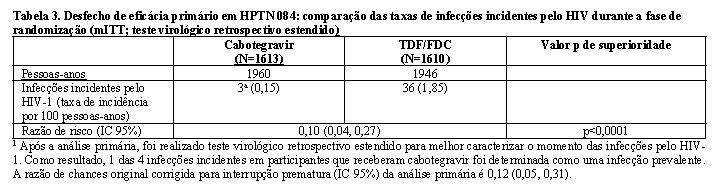
Estudo HPTN 084
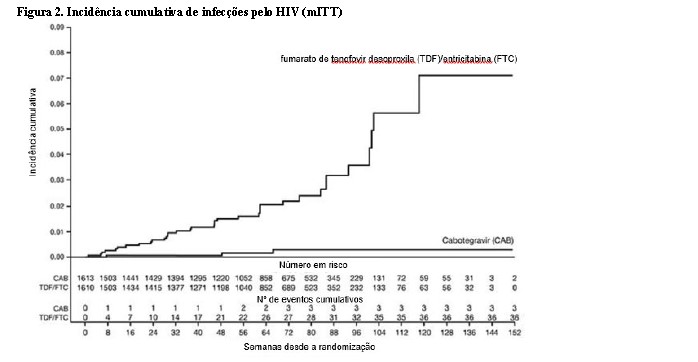
No estudo HPTN 084, de superioridade, 3.224 mulheres cisgênero foram randomizadas 1:1 e receberam cabotegravir (n = 1614) ou TDF/FTC (n = 1610) como medicação do estudo cego até a Semana 153. Na visita basal, a idade mediana dos participantes era de 25 anos, > 99% eram negros, > 99% eram mulheres cisgênero e 49% tinham < 25 anos de idade. O desfecho primário foi a taxa de infecções incidentes pelo HIV entre os participantes randomizados para cabotegravir oral e cabotegravir injetável em comparação com TDF/FTC oral. A análise primária demonstrou a superioridade de cabotegravir em comparação com TDF/FTC (corrigido para interrupção prematura) com uma redução de 88% no risco de adquirir a infecção incidente pelo HIV-1, razão de chances (IC 95%) 0,12 (0,05, 0,31); testes posteriores revelaram 1 das infecções em cabotegravir como prevalente, gerando 90% de redução no risco de infecção incidente por HIV-1 em relação ao TDF/FTC (consulte a Tabela 3).
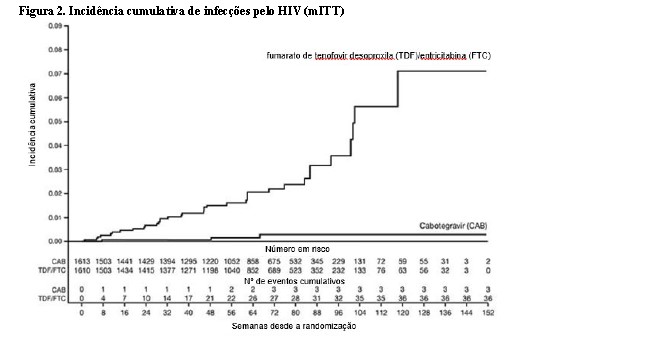
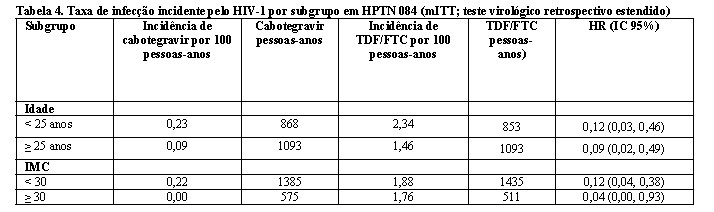
Os achados das análises de subgrupo pré-planejadas foram consistentes com o efeito protetor global, com uma taxa inferior de infecções incidentes por HIV-1 observada para participantes randomizados para o grupo cabotegravir em comparação com participantes randomizados para o grupo TDF/FTC (consulte Tabela 4).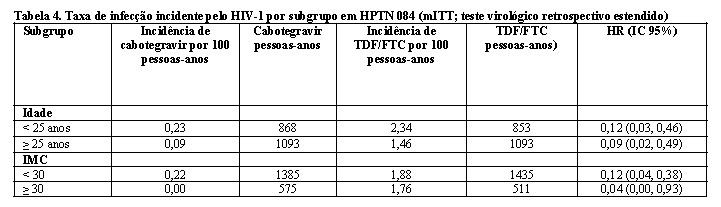
Estudo MOCHA (adolescentes infectados com HIV)
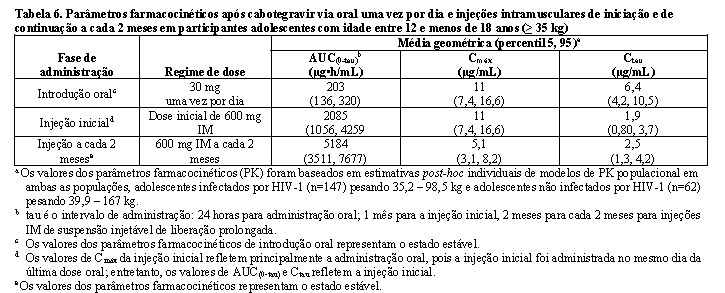
A segurança, tolerabilidade e farmacocinética de cabotegravir oral e injetável foram avaliadas em um estudo de fase I/II, multicêntrico, aberto, não comparativo, MOCHA (IMPAACT 2017, estudo 208580). 30 adolescentes infectados pelo HIV-1 e virologicamente suprimidos, com idade entre 12 e < 18 anos, pesando pelo menos 35 kg foram incluídos e receberam um comprimido de 30 mg de cabotegravir, diariamente, por 4 semanas, seguido por cabotegravir injetável mensal por 3 meses (mês 1: injeção de 600 mg, meses 2 e 3: injeção de 400 mg), ou por cabotegravir injetável a cada 2 meses durante 2 meses (mês 1 e 2: injeção de 600 mg), enquanto continua com o cART de base. Na visita basal, a mediana da idade dos participantes foi de 15,0 anos, o peso mediano foi de 47,9 kg, 47% eram do sexo feminino, 100% eram não brancos, nenhum participante tinha contagem de células CD4+ inferior a 350 células por mm3. Os desfechos primários na Semana 16 para os participantes com cabotegravir, que foram para confirmar as doses, segurança e farmacocinética de cabotegravir oral e injetável, em adolescentes infectados pelo HIV virologicamente suprimidos, foram atingidos. (ver Farmacocinética, Populações especiais, Reações adversas).
3. CARACTERÍSTICAS FARMACOLÓGICAS
Propriedades Farmacodinâmicas
Código ATC
Grupo farmacoterapêutico: antiviral para uso sistêmico, outros antivirais. Código ATC: J05AJ04
Mecanismo de ação
O cabotegravir inibe a integrase do HIV ligando-se ao local ativo da integrase e bloqueando a etapa de transferência da fita da integração do ácido desoxirribonucleico (DNA) retroviral que é essencial para o ciclo de replicação do HIV.
Efeitos farmacodinâmicos
Atividade antiviral em cultura celular
O cabotegravir exibiu atividade antiviral contra cepas de laboratório de HIV-1 do tipo selvagem com a concentração média de cabotegravir necessária para reduzir a replicação viral em 50 por cento (EC50) de 0,22 nM em células mononucleares do sangue periférico (PBMCs), 0,74 nM em células 293T e 0,57nM em células MT-4. O cabotegravir demonstrou atividade antiviral em cultura de células contra um painel de 24 isolados clínicos de HIV-1 (três em cada subtipos do Grupo M, incluindo A, B, C, D, E, F e G e 3 no grupo O) com valores de EC50 variando de 0,02 nM a 1,06 nM para HIV-1. Os valores de EC50 do cabotegravir contra três isolados clínicos de HIV-2 variaram de 0,10 nM a 0,14 nM. Não há dados clínicos disponíveis em pacientes com HIV-2.
Atividade antiviral em combinação com outros agentes antivirais
Nenhum fármaco com atividade anti-HIV inerente teve ação antagonista à atividade antirretroviral do cabotegravir (avaliações in vitro foram realizadas em combinação com rilpivirina, lamivudina, tenofovir e entricitabina).
Efeito do soro humano e das proteínas séricas
Estudos in vitro sugeriram um desvio de 408 vezes na IC50 do cabotegravir na presença de soro humano 100% (pelo método de extrapolação) e a IC50 ajustada das proteínas (PA-IC50) foi estimada em 102 nM em células MT4.
Resistência in vitro
Isolamento do HIV-1 do tipo selvagem e atividade contra cepas resistentes: vírus com aumento de > 10 vezes da EC50 do cabotegravir não foram observados durante a passagem de 112 dias da cepa IIIB. As seguintes mutações de integrase (IN) surgiram após a passagem do HIV-1 do tipo selvagem (com polimorfismo T124A) na presença de cabotegravir: Q146L (intervalo de aumento de 1,3-4,6), S153Y (intervalo de aumento de 2,8-8,4) e I162M (intervalo de aumento de = 2,8). Como observado acima, a detecção do T124A é a seleção de uma variante minoritária preexistente que não apresenta susceptibilidade diferencial ao cabotegravir. Nenhuma substituição de aminoácidos na região da integrase foi selecionada na passagem do HIV- 1 tipo selvagem NL-432 na presença de 6,4 nM de cabotegravir até o Dia 56. Entre os múltiplos mutantes, a maior variação foi observada nas mutantes contendo Q148K ou Q148R. E138K/Q148H resultou em uma redução de 0,92 vezes na suscetibilidade ao cabotegravir, mas a E138K/Q148K resultou em uma redução de 81 vezes na suscetibilidade ao cabotegravir. G140C/Q148R e G140S/Q148R resultaram em uma redução de 22 e de 12 vezes na suscetibilidade ao cabotegravir, respectivamente. Embora a N155H não tenha alterado a suscetibilidade ao cabotegravir, a N155H/Q148R resultou em uma redução de 61 vezes na suscetibilidade ao cabotegravir.
Resistência in vivo
Estudo HPTN 083
Na análise primária do estudo HPTN 083, houve 13 infecções incidentes no braço cabotegravir e 39 infecções incidentes no braço fumarato de tenofovir desoproxila (TDF)/entricitabina (FTC). No braço cabotegravir, ocorreram 5 infecções incidentes ao receber injeções de cabotegravir como profilaxia pré-exposição, dos quais 4 participantes receberam injeções no prazo e 1 participante recebeu uma injeção fora do cronograma. Cinco infecções incidentes ocorreram ≥ 6 meses após a última dose de cabotegravir como profilaxia pré-exposição. Três infecções incidentes ocorreram durante o período de introdução oral. Tentou-se realizar a genotipagem e fenotipagem de HIV na primeira visita onde a carga viral de HIV era > 500 cópias/mL. Das 13 infecções incidentes no braço cabotegravir, 4 participantes tinham mutações de resistência a INSTI. No braço TDF/FTC, os 4 participantes com resistência a NRTI (incluindo 3 que tinham resistência a múltiplas classes) incluíram 3 com M184V/I e um com K65R. Nenhum dos 5 participantes que foram infectados após interrupção prolongada da administração de cabotegravir apresentava mutações de resistência a INSTI. Não foi possível gerar nem o genótipo nem o fenótipo para um dos 5 participantes, com apenas 770 cópias/mL de RNA de HIV-1. O fenótipo da integrase não pôde ser gerado para um dos 4 participantes restantes. Os 3 participantes restantes mantiveram a suscetibilidade a todos INSTIs. Três participantes se infectaram durante a fase de introdução oral, antes de receber cabotegravir injetável. Um participante com níveis plasmáticos indetectáveis de cabotegravir não apresentava mutações de resistência a INSTI e era suscetível a todos INSTIs. Dois participantes com concentrações detectáveis de cabotegravir no plasma apresentaram mutações de resistência a INSTI. O primeiro participante tinha mutações resistentes a INSTI E138E/K, G140G/S, Q148R e E157Q. Não pôde ser gerado o fenótipo da integrase. O segundo participante tinha mutações de resistência a INSTI E138A e Q148R. Este vírus era resistente ao cabotegravir (intervalo de aumento = 5,92), mas suscetível à dolutegravir (intervalo de aumento = 1,69). Cinco participantes adquiriram HIV-1, apesar das injeções de cabotegravir no prazo para 4 participantes e uma injeção fora do cronograma para um participante. Dois participantes apresentavam cargas virais muito baixas para serem analisadas. O terceiro participante não tinha mutações de resistência a INSTI na primeira visita virêmica (Semana 17), mas tinha R263K em 112 e 117 dias depois. Embora o fenótipo não possa ser determinado 112 dias depois, o fenótipo do dia 117 mostrou que esse vírus é suscetível ao cabotegravir (intervalo de aumento = 2,32) e ao dolutegravir (intervalo de aumento = 2,29). O quarto participante tinha mutações de resistência a INSTI G140A e Q148R. O fenótipo mostrou resistência ao cabotegravir (intervalo de aumento = 13), mas suscetibilidade ao dolutegravir (intervalo de aumento = 2,09). O quinto participante não tinha mutações de resistência a INSTI. Além das 13 infecções incidentes, um outro participante estava infectado com HIV-1 no momento da inclusão e não apresentava mutações de resistência a INSTI naquele momento; no entanto, 60 dias depois, a mutação de resistência a INSTI E138K e Q148K foram detectadas. O fenótipo não pôde ser gerado. Seguindo a análise primária, um teste virológico retrospectivo estendido foi realizado para melhor caracterizar o momento das infecções por HIV. Como resultado, uma das 13 infecções incidentes em um participante que recebeu dentro do prazo cabotegravir injetável foi considerada uma infecção prevalente.
Estudo HPTN 084
Na análise primária do estudo HPTN 084, houve 4 infecções incidentes no braço cabotegravir e 36 infecções incidentes no braço TDF/FTC. No braço cabotegravir, ocorreram 2 infecções incidentes durante a administração de injeções; um participante apresentava 3 injeções atrasadas de cabotegravir e ambos eram não aderentes ao cabotegravir oral. Duas infecções incidentes ocorreram após a última dose de cabotegravir oral; ambos os participantes não eram aderentes ao cabotegravir oral. A primeira visita HIV positiva ocorreu aprox. 11 semanas após a inclusão para um participante e 57 semanas após a inclusão para o outro. A genotipagem de HIV foi tentada na primeira visita onde a carga viral de HIV era > 500 c/mL (primeira visita virêmica). Os resultados da genotipagem de HIV estavam disponíveis para 3 dos 4 participantes do braço cabotegravir. Nenhuma mutação importante de resistência a INSTI foi detectada. Os resultados da genotipagem de HIV estavam disponíveis para 33 das 36 infecções incidentes no grupo TDF/FTC. Um participante tinha uma mutação principal em NRTI (M184V); este participante também apresentava resistência a NNRTI com a mutação K103N. Nove outros participantes tinham resistência a NNRTI (7 apresentavam K103N, isolado ou com E138A ou P225H; 1 apresentava K101E isolado; 1 apresentava E138A isolado). Seguindo a análise primária, teste virológico retrospectivo estendido foi realizado para melhor caracterizar o momento das infecções por HIV
1. Como resultado, 1 das 4 infecções incidentes por HIV-1 em participantes que receberam cabotegravir foi determinada como infecção prevalente.
Estudo HPTN 083-01 e HPTN 084-01
Nos estudos HPTN 083-01 e HTPN 084-01, não foram observadas infecções incidentes entre 64 adolescentes em risco (pesando 35 kg ou mais) recebendo cabotegravir para prevenção de HIV-1.
Efeitos no eletrocardiograma
Em um estudo randomizado, controlado por placebo, cruzado (cross-over), de três períodos, 42 indivíduos saudáveis foram randomizados em 6 sequências aleatórias e receberam três doses de administração oral de placebo, cabotegravir 150 mg a cada 12 horas (Cmáx média no estado de equilíbrio foi aproximadamente 2,8 vezes e 5,6 vezes acima da dose oral uma vez ao dia de 30 mg cabotegravir comprimidos revestidos e de 600 mg a cada 2 meses de cabotegravir injetável, respectivamente) ou dose única de 400 mg de moxifloxacino (controle ativo). Após o ajuste basal e do placebo, a alteração máxima pareada pelo tempo na média do QTc baseada no método de correção de Fridericia (QTcF) para
o cabotegravir foi de 2,62 ms (IC superior de 90% unilateral: 5,26 ms). O cabotegravir não prolongou o intervalo QTc durante 24 horas pósdose.
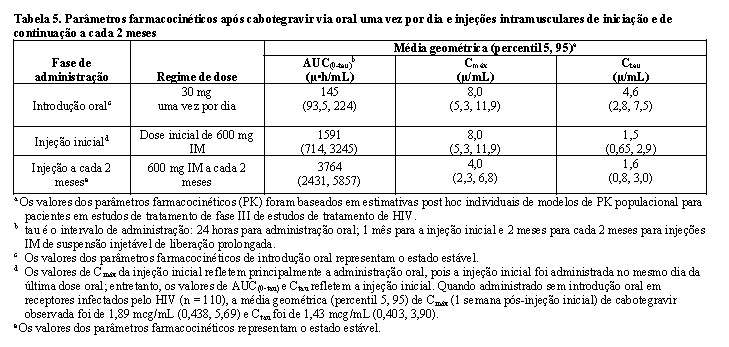
Propriedades Farmacocinéticas
A farmacocinética (PK) do cabotegravir é semelhante entre indivíduos saudáveis e os vivendo com HIV. A variabilidade PK do cabotegravir é moderada a alta. Nos estudos de Fase I em indivíduos saudáveis, o % do CVb entre indivíduos para a AUC, Cmáx e Ctau variou de 34 a 91% em estudos com indivíduos saudáveis. A variabilidade intra indivíduos (% CVw) é menor que a variabilidade entre indivíduos.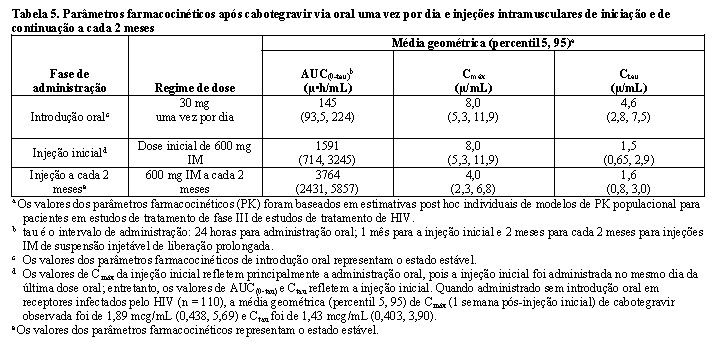
Absorção
O cabotegravir é rapidamente absorvido após administração oral, com Tmáx médio de 3 horas após a dose para a formulação em comprimidos. A linearidade da farmacocinética do cabotegravir é dependente da dose e da formulação. Após a administração oral das formulações de comprimidos, a farmacocinética do cabotegravir foi proporcional à dose e ligeiramente menor que proporcional à dose de 5 mg a 60 mg. Com uma dose de uma vez ao dia, o estado de equilíbrio farmacocinético é alcançado em 7 dias. O cabotegravir pode ser administrado com ou sem alimentos. Os alimentos aumentaram a extensão da absorção de cabotegravir. A biodisponibilidade do cabotegravir é independente do conteúdo da refeição: refeições com alto teor de gordura aumentaram a AUC(0-∞) do cabotegravir em 14% e aumentaram a Cmáx em 14% em relação às condições de jejum. Esses aumentos não são clinicamente significativos. A biodisponibilidade absoluta do cabotegravir não foi estabelecida.
Distribuição
O cabotegravir é altamente ligado (aproximadamente > 99%) às proteínas plasmáticas humanas, com base em dados in vitro. Após a administração de comprimidos orais, o volume de distribuição aparente em fase terminal (Vz/F) no plasma foi de 12,3 L (conforme resultados obtidos no estudo 205696 ministrado em jejum com comprimidos orais de 30 mg). Nos humanos, o volume distribuição aparente do compartimento central (Vc/F) estimado do cabotegravir plasmático foi de 5,27 L e o volume de distribuição aparente do compartimento periférico (Vp/F) foi de 2,43 L. Essas estimativas de volume, juntamente com suposição de alta biodisponibilidade (F), sugerem alguma distribuição do cabotegravir para o espaço extracelular. O cabotegravir está presente no trato genital feminino e masculino, após uma injeção IM única de 3 mL (600 mg), conforme observado em um estudo em participantes saudáveis (n = 15). As concentrações medianas de cabotegravir no Dia 3 (a primeira amostra tecidual de PK) foram de 0,49 mg/mL no tecido cervical, 0,29 mg/mL no fluido cervicovaginal, 0,37 mg/mL no tecido vaginal, 0,32 mg/mL no tecido retal, e 0,69 mg/mL no fluido retal, que estão acima de PA-IC90
in vitro.
Metabolismo
O cabotegravir é principalmente metabolizado pela UGT1A1 com um componente UGT1A9 menor. O cabotegravir é o composto circulante predominante no plasma, representando > 90% do radiocarbono total no plasma. Após administração oral em humanos, o cabotegravir é eliminado principalmente pelo metabolismo; a eliminação renal do cabotegravir inalterado é baixa ( < 1% da dose). Quarenta e sete por cento da dose oral total são excretados como cabotegravir inalterado nas fezes. Não se sabe se tudo ou parte disso se deve ao fármaco não absorvido ou excreção biliar do conjugado glicuronizado, que pode ser degradado posteriormente para formar o composto original no lúmen intestinal. Observou-se que o cabotegravir pode estar presente em amostras de bile duodenal. O metabólito do ácido glicurônico também estava presente em algumas, mas não todas as amostras de bile duodenal. Vinte e sete por cento da dose oral total é excretada na urina, principalmente como um metabólito de glicuronídeo (75% da radioatividade da urina, 20% da dose total).
Eliminação
O cabotegravir tem uma meia-vida terminal média de 41 h e um clearance aparente (CL/F) de 0,21 L por hora, com base em análises farmacocinéticas populacionais.
Populações especiais de pacientes
Gênero
As análises farmacocinéticas populacionais não revelaram efeito clinicamente relevante do gênero na exposição ao cabotegravir. Além disso, não foram observadas diferenças clinicamente relevantes nas concentrações plasmáticas de cabotegravir no estudo HPTN 083 por sexo, incluindo em homens cisgênero e mulheres transgênero com ou sem uso de terapia hormonal de sexo cruzado. Portanto, nenhum ajuste de dose é necessário com base no sexo.
Raça
As análises farmacocinéticas populacionais não revelaram efeito clinicamente relevante da raça na exposição ao cabotegravir, portanto, nenhum ajuste de dose é necessário com base na raça.
IMC
As análises farmacocinéticas populacionais não revelaram efeito clinicamente relevante do IMC na exposição ao cabotegravir, portanto, nenhum ajuste de dose é necessário com base no IMC.
Adolescentes
As análises farmacocinéticas populacionais não revelaram diferenças clinicamente relevantes na exposição entre os participantes adolescentes e os participantes adultos infectados e não infectados pelo HIV-1 do programa de desenvolvimento de cabotegravir, portanto, nenhum ajuste de dose é necessário para adolescentes com peso ≥ 35 kg.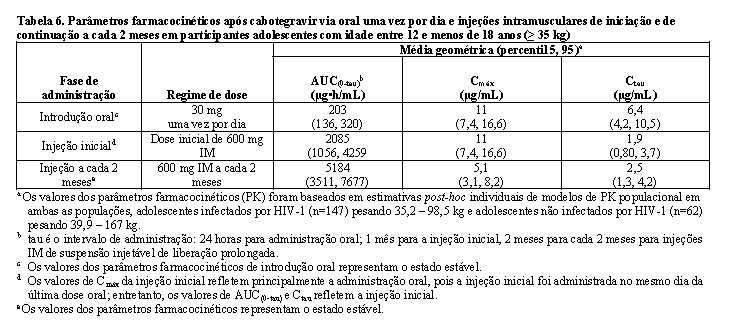
Crianças
A farmacocinética e as recomendações de administração de cabotegravir em indivíduos com menos de 12 anos de idade ou com menos de 35 kg não foram estabelecidas.
Idosos
A análise farmacocinética populacional do cabotegravir não revelou efeito clinicamente relevante da idade na exposição ao cabotegravir. Os dados farmacocinéticos do cabotegravir em indivíduos com idade > 65 anos são limitados.
Insuficiência renal
Não foram observadas diferenças farmacocinéticas clinicamente importantes entre indivíduos com insuficiência renal grave (CrCL < 30 mL/min e não dialíticos) e indivíduos saudáveis pareados. Nenhum ajuste de dose é necessário em pacientes com insuficiência renal leve (CL creatinina ≥60mL/min a < 90mL/min), moderada (CL creatinina ≥30mL/min a 60mL/min) ou grave (CL creatinina ≥15mL/min a < 30mL/min e não dialítico). O cabotegravir não foi estudado em pacientes em diálise.
Insuficiência hepática
Não foram observadas diferenças farmacocinéticas clinicamente importantes entre indivíduos com insuficiência hepática moderada e indivíduos saudáveis pareados. Nenhum ajuste de dose é necessário para pacientes com insuficiência hepática leve a moderada (pontuação de Child-Pugh A ou B). O efeito da insuficiência hepática grave (pontuação de Child-Pugh C) na farmacocinética do cabotegravir não foi estudado.
Coinfecção por hepatite B ou C
Não existem dados para o uso de cabotegravir como profilaxia pré-exposição ao vírus HIV-1 em indivíduos com infecção por vírus de hepatite B e C.
Polimorfismos em enzimas metabolizadoras de fármacos
Em uma metanálise de indivíduos saudáveis e vivendo com HIV, indivíduos vivendo com HIV com genótipos UGT1A1 que conferem um metabolismo pobre do cabotegravir tiveram um aumento de 1,2 vezes na AUC, Cmáx e Ctau médias do cabotegravir no estado de equilíbrio após administração de cabotegravir injetável vs. aumento médio de 1,38 vezes após a administração oral de cabotegravir. Isso foi semelhante ao aumento médio de 1,3 a 1,5 vezes no estado de equilíbrio do cabotegravir, AUC, Cmáx e Ctau do cabotegravir observado após cabotegravir oral em indivíduos saudáveis e vivendo com HIV combinados. Essas diferenças não são consideradas clinicamente relevantes. Polimorfismos em UGT1A9 não foram associados a diferenças na farmacocinética do cabotegravir, portanto, não é necessário ajuste de dose em indivíduos com polimorfismos em UGT1A1 ou UGT1A9.
4. CONTRAINDICAÇÕES
Apretude® é contraindicado para pacientes:
- com hipersensibilidade conhecida ao cabotegravir ou a qualquer um dos excipientes dos comprimidos;
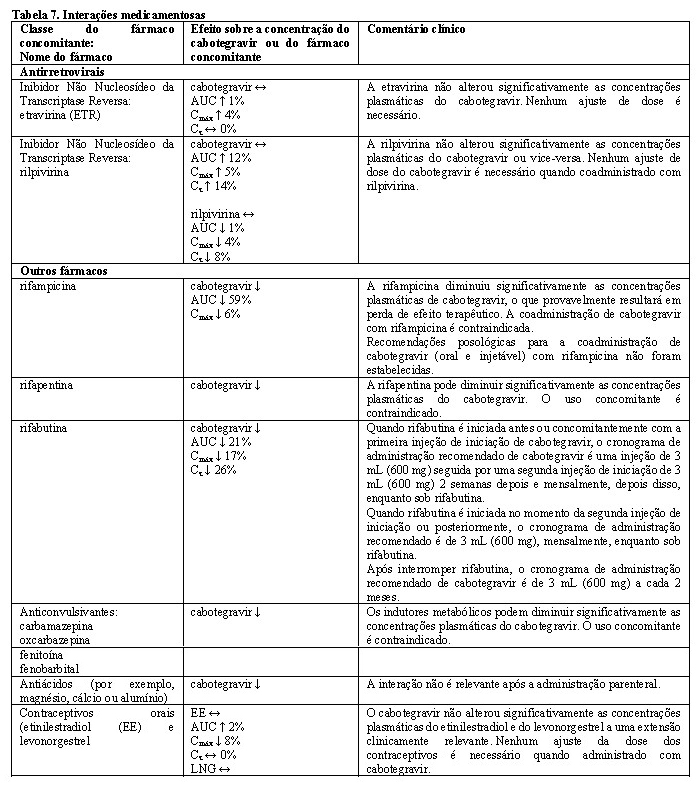
- recebendo rifampicina, rifapentina, fenitoína, fenobarbital, carbamazepina, oxcarbazepina ou outros medicamentos indutores fortes da enzima UGT1A1 (ver Interações medicamentosas);
- com resultado desconhecido ou positivo para HIV-1(ver Advertências e Precauções).
5. ADVERTÊNCIAS E PRECAUÇÕES
Apretude® nem sempre será eficaz na prevenção da aquisição de HIV-1 (ver Estudos clínicos). O momento para o início da proteção após o início de cabotegravir é desconhecido. Apretude® deve ser usado para profilaxia pré-exposição como parte de uma estratégia global de prevenção de infecção pelo HIV-1, incluindo
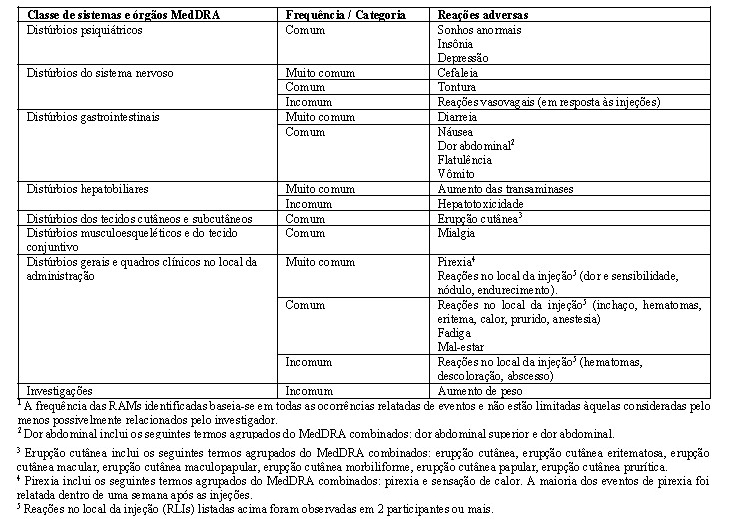
o uso de outras medidas de prevenção de HIV-1 (por exemplo, conhecimento do status de HIV-1, teste regular para outras infecções sexualmente transmissíveis, uso de preservativo). Apretude® só deve ser usado para reduzir o risco de adquirir HIV-1 em indivíduos confirmados como HIV negativos (ver Contraindicações). Os indivíduos devem ser reconfirmados como HIV negativos em intervalos frequentes (por exemplo, de acordo com as diretrizes locais, mas em intervalos de no máximo 3 meses) enquanto administram Apretude® para profilaxia pré-exposição. Se houver sintomas clínicos consistentes com infecção viral aguda e suspeita de exposições recentes ( < 1 mês) ao HIV-1, o status de HIV-1 deve ser reconfirmado. Foi relatada depressão com o uso de Apretude® (ver Reações Adversas). Os profissionais de saúde devem monitorar de perto os indivíduos com depressão ou comportamento suicida para avaliar se os sintomas estão relacionados ao Apretude® e determinar se os riscos do uso continuado do Apretude® superam os benefícios.
Risco potencial de resistência
Há um risco potencial de desenvolver resistência ao cabotegravir se um indivíduo adquirir HIV-1 antes ou durante a administração de cabotegravir, ou após a descontinuação da profilaxia pré-exposição com cabotegravir. Para minimizar isso, é essencial reavaliar clinicamente os indivíduos quanto ao risco de aquisição de HIV e realizar testes frequentes para confirmar o status HIV negativo. Indivíduos com suspeita ou confirmação de HIV-1 devem iniciar imediatamente a terapia antirretroviral. Formas alternativas de profilaxia pré-exposição devem ser consideradas após a descontinuação de cabotegravir para aqueles indivíduos com risco contínuo de aquisição de HIV e iniciadas dentro de 2 meses após a injeção final de cabotegravir.
Propriedades da ação prolongada de cabotegravir injetável
As concentrações residuais de cabotegravir injetável podem permanecer na circulação sistêmica dos indivíduos por períodos prolongados (até 12 meses ou mais), portanto, os médicos devem levar em consideração as características de liberação prolongada de cabotegravir quando o medicamento for descontinuado (ver Interações, Gravidez e Lactação e superdosagem).
Importância da adesão
Os indivíduos devem ser aconselhados periodicamente a cumprir estritamente o cronograma de administração recomendado de cabotegravir, a fim de reduzir o risco de aquisição de HIV-1 e o desenvolvimento potencial de resistência.
Reações de hipersensibilidade
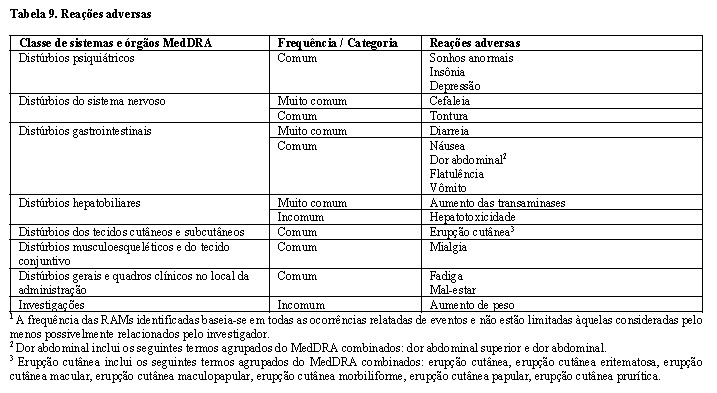
Foram relatadas reações de hipersensibilidade associadas a inibidores de integrase. Essas reações foram caracterizadas por erupção cutânea, achados constitucionais e, algumas vezes, disfunção orgânica, incluindo lesão hepática. Deve-se interromper imediatamente o cabotegravir e outros agentes suspeitos, caso sinais ou sintomas de hipersensibilidade sejam desenvolvidos (incluindo, mas não se limitando a, erupção cutânea grave ou erupção cutânea acompanhada de febre, mal-estar geral, fadiga, dores musculares ou articulares, bolhas, lesões orais, conjuntivite, edema facial, hepatite, eosinofilia ou angioedema). O estado clínico, incluindo aminotransferases hepáticas, deve ser monitorado e terapia apropriada deve ser iniciada. (ver Posologia e Modo de Usar, Contraindicações, Reações Adversas e Estudos Clínicos)
Hepatotoxicidade
Foi relatada hepatotoxicidade em um número limitado de pacientes recebendo cabotegravir com ou sem doença hepática preexistente conhecida (ver Reações adversas). O monitoramento clínico e laboratorial é recomendado e o tratamento com cabotegravir deve ser descontinuado se a hepatotoxicidade for confirmada e os indivíduos tratados de acordo com a indicação clínica.
Interações medicamentosas
Deve-se ter cuidado ao prescrever cabotegravir com medicamentos que possam reduzir sua exposição (ver Interações Medicamentosas).
Gravidez e Lactação
Fertilidade
Estudos em animais indicam que não há efeitos do cabotegravir na fertilidade masculina ou feminina (ver Informações Não Clínicas, abaixo).
Gravidez
Existem dados limitados para cabotegravir em mulheres grávidas. O efeito na gravidez humana é desconhecido. O cabotegravir não foi teratogênico quando estudado em ratas e coelhas prenhes, mas causou um atraso no parto que foi associado à redução da sobrevida e viabilidade da prole de ratos em exposições superiores às doses terapêuticas (ver Informações Não Clínicas, abaixo). A relevância para a gravidez humana é desconhecida. O cabotegravir deve ser utilizado durante a gestação apenas se o benefício esperado justificar o potencial risco ao feto. O cabotegravir foi detectado na circulação sistêmica por até 12 meses ou mais após a uma injeção, portanto, deve-se considerar o potencial de exposição ao feto durante a gravidez (ver Advertências e Precauções - Propriedades da ação prolongada de cabotegravir injetável).
Lactação
Espera-se que o cabotegravir seja secretado no leite humano com base em dados com animais, embora isso não tenha sido confirmado em humanos. O cabotegravir pode estar presente no leite humano por até 12 meses ou mais após a última administração de cabotegravir injetável. Recomenda-se a administração de cabotegravir em lactantes apenas se o benefício esperado justificar o risco potencial para o bebê.
Categoria C de risco na gravidez. Este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica ou do cirurgião-dentista.
Efeitos sobre a capacidade de dirigir veículos e operar máquinas
Não houve estudos para investigar o efeito do cabotegravir sobre a capacidade de dirigir ou operar máquinas. É preciso levar em conta o estado clínico do paciente e o perfil de eventos adversos de cabotegravir ao avaliar essa capacidade.
Informações Não-Clínicas
Carcinogênese/Mutagênese
O cabotegravir não foi mutagênico ou clastogênico com uso de testes in vitro em bactérias e cultura de células de mamíferos, e nem em teste do micronúcleo in vivo em ratos. O cabotegravir não foi carcinogênico em estudos em longo prazo em camundongos e ratos.
Toxicologia Reprodutiva
Fertilidade
O cabotegravir quando administrado por via oral em ratos machos e fêmeas a 1000 mg/kg/dia ( > 30 vezes a exposição em humanos na Dose Máxima Recomendada para Humanos [MRHD] de 30 mg por via oral ou na dose de 400 mg IM) por até 26 semanas não causou efeitos adversos nos órgãos reprodutores dos machos ou fêmeas ou na espermatogênese. Não foi observado nenhum efeito funcional no acasalamento ou na fertilidade dos machos ou fêmeas em ratos que receberam cabotegravir em doses de até 1000 mg/kg/dia.
Gravidez
Em um estudo de desenvolvimento embriofetal, não houve desfechos adversos no desenvolvimento após a administração oral de cabotegravir à coelhas prenhes em doses até 2000mg/kg/dia (0,66 vezes a exposição em humanos na MRHD de 30 mg por via oral ou aproximadamente 1 vezes a dose IM de 400 mg) ou a ratas prenhes em doses de até 1000 mg/kg/dia ( > 30 vezes a exposição em humanos na MRHD de 30 mg por via oral ou a dose de IM de 400 mg). Em ratos, alterações no crescimento fetal (diminuição do peso corporal) na ausência de toxicidade materna foi observada em 1000 mg/kg/dia. Estudos em ratas prenhes mostraram que o cabotegravir atravessa a placenta e pode ser detectado no tecido fetal. Dados não clínicos de estudos pré e pós-natal (PPN) em ratos com 1.000 mg/kg/dia ( > 30 vezes a exposição em humanos na MRHD de 30 mg por via oral ou na dose IM de 400 mg), o cabotegravir atrasou o início do parto e, em algumas ratas, esse atraso foi associado com um maior número de natimortos e mortalidade neonatal imediatamente após o nascimento. Uma dose mais baixa de 5 mg/kg/dia de cabotegravir ( > 10 vezes a exposição em humanos na MRHD de 30 mg por via oral ou na dose IM de 400 mg) não se associou ao atraso no parto ou à mortalidade em ratos. Em estudos com coelhos e ratos, não houve efeito na sobrevida quando os fetos foram paridos por cesariana. Quando filhotes de ratos nascidos de mães tratadas com cabotegravir foram adotados ao nascimento e amamentados por mães controle, incidências similares de mortalidade foram observadas.
Toxicologia e/ou farmacologia animal
O efeito do tratamento diário prolongado com doses elevadas de cabotegravir tem sido avaliado em estudos de toxicidade de dose oral repetida em ratos (26 semanas) e em macacos (39 semanas). Não houve efeitos adversos relacionados ao fármaco em ratos ou macacos que receberam cabotegravir por via oral em doses de até 1000 mg/kg/dia ou 500 mg/kg/dia, respectivamente. No estudo de toxicidade de 14 dias em macacos, uma dose de 1000 mg/kg/dia não foi tolerada e resultou em morbidade associada a efeitos gastrointestinais (GI) (perda de peso corporal, vômitos, fezes soltas/aquosas e desidratação moderada a grave).
No estudo de toxicidade de 28 dias em macacos, a exposição no final do estudo a 500 mg/kg/dia foi semelhante à alcançada no estudo de 14 dias a 1000 mg/kg/dia. Isso sugere que a intolerância gastrointestinal observada no estudo de 14 dias foi o resultado da administração local de medicamentos e não toxicidade sistêmica. Em um estudo de 3 meses em ratos, quando o cabotegravir foi administrado por injeção subcutânea (SC) (até 100 mg/kg/dose); injeção IM mensal (até 75 mg/kg/dose) ou injeção SC semanal (100 mg/kg/dose), não houve efeitos adversos observados e nenhuma nova toxicidade para órgãos-alvo (em exposições > 30 vezes a exposição em humanos na MRHD da dose IM de 400 mg).
Populações Especiais
Ver Populações Especiais de Pacientes em Características Farmacológicas.
6. INTERAÇÕES MEDICAMENTOSAS
Efeito do cabotegravir na farmacocinética de outros agentes
In vivo, o cabotegravir não teve efeito sobre o midazolam, um substrato da CYP3A4. O cabotegravir não é um inibidor clinicamente relevante das seguintes enzimas e transportadores: CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP3A4, UGT1A1, UGT1A3, UGT1A4, UGT1A6, UGT1A9, UGT2B4, UGT2B7, UGT2B15 e UGT2B17, gp-P, proteína de resistência do câncer de mama (BCRP), bomba de exportação de sais biliares (BSEP), transportador de cátions orgânicos (OCT) 1, OCT2, OATP1B1, OATP1B3, proteína transportadora do sistema de efluxo de múltiplos fármacos e toxinas (MATE) 1, MATE 2-K, proteína de resistência a múltiplas drogas (MRP) 2 ou MRP4.
O cabotegravir inibiu in vitro os transportadores de ânions orgânicos (OAT) 1 (IC50 = 0,81 mM) e OAT3 (IC50 = 0,41 mM), no entanto, na modelagem farmacocinética de base fisiológica (PBPK), interação com substratos OAT em concentrações clinicamente relevantes não é esperada. In vitro, o cabotegravir não induziu CYP1A2, CYP2B6 ou CYP3A4. Com base nesses dados e nos resultados de estudos de interação medicamentosa, não é esperado que o cabotegravir afete a farmacocinética dos fármacos que são substratos dessas enzimas ou transportadores. Com base no perfil in vitro e clínico de interação medicamentosa, não é esperado que o cabotegravir altere as concentrações de outros medicamentos antirretrovirais, incluindo inibidores de protease, inibidores nucleosídeos da transcriptase reversa, inibidores não nucleosídeos da transcriptase reversa, inibidores da integrase, inibidores de entrada e ibalizumabe.
Efeito de outros agentes na farmacocinética do cabotegravir
O cabotegravir é metabolizado principalmente pelo UGT1A1 com alguma contribuição do UGT1A9. Espera-se que os medicamentos que são fortes indutores de UGT1A1 ou UGT1A9 diminuam as concentrações de plasma de cabotegravir levando à falta de eficácia (ver Contraindicações). Simulações usando o modelo PBPK mostram que nenhuma interação clinicamente significativa é esperada após a administração concomitante de cabotegravir com fármacos que inibem as enzimas UGT. In vitro, o cabotegravir não foi um substrato do OATP1B1, OATP1B3, OATP2B1 ou OCT1. O cabotegravir é um substrato da gp-P e da BCRP, no entanto, devido à sua alta permeabilidade, não se espera nenhuma alteração na absorção quando coadministrado com inibidores da gp-P ou BCRP. Não foram realizados estudos de interação medicamentosa com cabotegravir injetável. Os dados de interação do fármaco fornecidos na Tabela 7 são obtidos a partir de estudos com cabotegravir oral.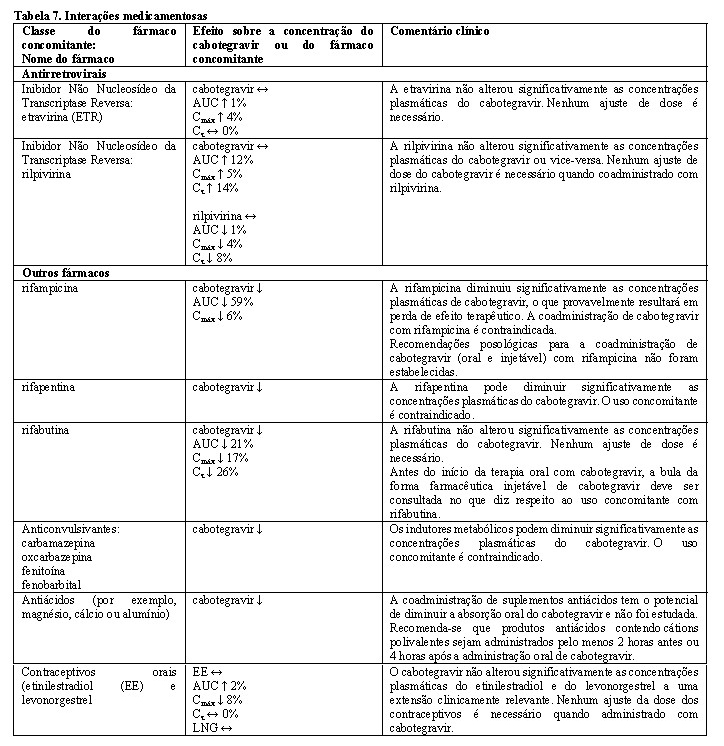
7. CUIDADOS DE ARMAZENAMENTO DO MEDICAMENTO
Cuidados de Armazenamento
Mantenha o produto na embalagem original e em temperatura ambiente (entre 15°C e 30°C). O prazo de validade é de 60 meses a partir da data de fabricação, impressa na embalagem externa do produto.
Número de lote e datas de fabricação e validade: vide embalagem.
Não use medicamento com o prazo de validade vencido. Guarde-o em sua embalagem original.
Aspectos físicos / Características organolépticas
Comprimido revestido oval, de cor branca, gravado com 'SV CTV' em um lado.
Antes de usar, observe o aspecto do medicamento.
Todo medicamento deve ser mantido fora do alcance das crianças.
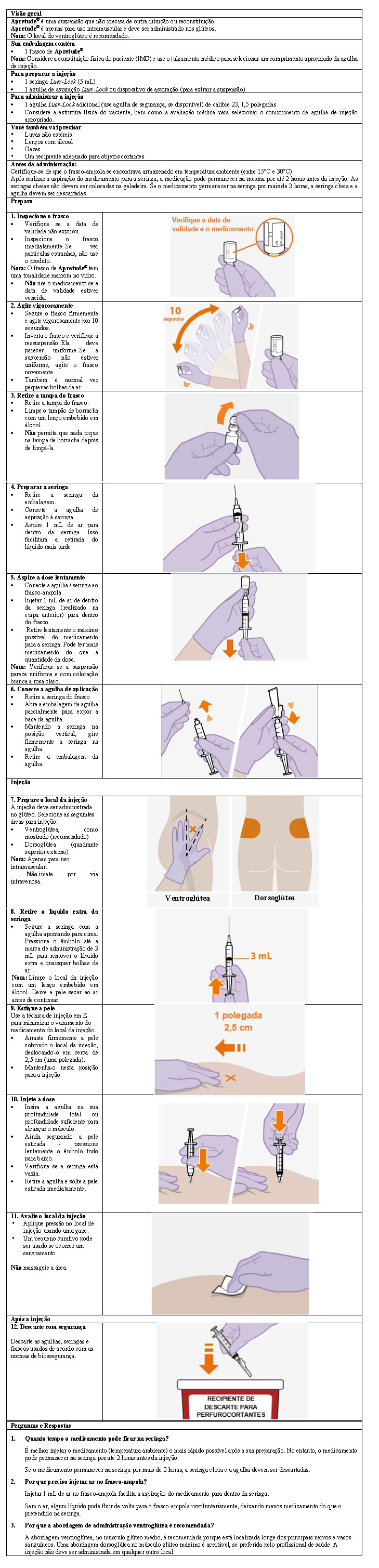
8. POSOLOGIA E MODO DE USAR
Posologia
A terapia deverá ser iniciada por um médico com experiência no tratamento e prevenção da infecção por HIV. Antes de iniciar a utilização do Apretude®, os indivíduos devem ter um teste de HIV-1 negativo documentado, de acordo com as diretrizes apli