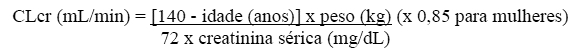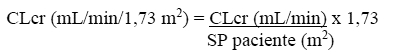ANTARA
EUROFARMA
levetiracetam
Antiepiléptico.
Apresentações.
Comprimido revestido de 250 mg ou 750 mg: embalagem com 30 comprimidos revestidos.
USO ORAL
USO ADULTO E PEDIÁTRICO ACIMA DE 6 ANOS
Composição.
Cada comprimido revestido de 250 mg contém: levetiracetam 250 mg; excipientes* q.s.p 1 comprimido revestido. *Excipientes: croscarmelose sódica, dióxido de silício, copovidona, macrogol, estearato de magnésio, álcool polivinílico, talco, dióxido de titânio e óxido de ferro amarelo.
Cada comprimido revestido de 750 mg contém: levetiracetam 750 mg; excipientes* q.s.p 1 comprimido revestido. *Excipientes: croscarmelose sódica, dióxido de silício, copovidona, macrogol, estearato de magnésio, álcool polivinílico, talco, dióxido de titânio, amarelo de quinolina, sacarose, amarelo crepúsculo, cloroidróxido de alumínio.
Informações técnicas.
1. INDICAÇÕES
Este medicamento é indicado como monoterapia para o tratamento de crises focais/parciais, com ou sem generalização secundária em pacientes a partir dos 16 anos com diagnóstico recente de epilepsia.
Antara (levetiracetam) é indicado como terapia adjuvante no tratamento de:
- Crises focais/parciais com ou sem generalização secundária em adultos, adolescentes e crianças com idade superior a 6 anos, com epilepsia;
- Crises mioclônicas em adultos, adolescentes e crianças com idade superior a 12 anos, com epilepsia mioclônica juvenil;
- Crises tônico-clônicas primárias generalizadas em adultos, adolescentes e crianças com mais de 6 anos de idade, com epilepsia idiopática generalizada.
2. RESULTADOS DE EFICÁCIA
Crises focais/parciais em adultos e adolescentes a partir de 16 anos com epilepsia
- Monoterapia:
O estudo duplo-cego, com grupo paralelo, de não inferioridade, para monoterapia foi realizado comparando-se o levetiracetam (LEV) e carbamazepina (CBZ) de liberação controlada em pacientes de 16 anos de idade ou acima com diagnóstico de epilepsia de início recente. As crises foram focais/parciais não provocadas (tipo IA, IB ou IC com clara origem focal) ou crises tônico-clônicas generalizadas (sem clara origem focal), categorizadas de acordo com a Classificação Internacional das Crises e Epilepsias da Liga Internacional Contra a Epilepsia (ILAE). O estudo foi realizado em 85 centros de 13 países (Europa e África do Sul).
Ao final do período de triagem de 1 semana, pacientes elegíveis foram estratificados pelo tipo de crise (IA/IB/IC ou IC/IIE sem clara origem focal) e randomicamente atribuídos para receber CBZ CR (n = 291) ou LEV (n = 285), por até 121 semanas dependendo da resposta. Conservadoramente, uma formulação de liberação controlada (CR) de carbamazepina foi utilizada para minimizar os eventos adversos.
O tratamento foi iniciado com uma titulação de 2 semanas tanto com carbamazepina CR 200 mg/dia ou levetiracetam 500 mg/dia, seguido por uma estabilização de 1 semana nos níveis da dose alvo (carbamazepina CR 400 mg/dia ou levetiracetam 1000 mg/dia).
Os pacientes que não apresentaram crises durante o período de avaliação de 26 semanas permaneceram com esta dose neste período, e nas 26 semanas seguintes com terapia de manutenção. Se um paciente tivesse uma crise durante o período de avaliação, um escalonamento (feito ao longo de 2 semanas com estabilização de 1 semana) para a dose de nível 2 deveria ser feita (carbamazepina CR 800 mg/dia ou levetiracetam 2000 mg/dia). De modo similar, os pacientes que tiveram uma crise durante o período de avaliação da dose de nível 2 puderam passar por outro escalonamento de dose para carbamazepina 1200 mg/dia ou levetiracetam 3000 mg/dia.
Nos níveis de dose 2 e 3, o período de avaliação foi constituído por 26 semanas, seguido por um período de manutenção de 26 semanas.
Quinhentos e setenta e nove (579) pacientes foram randomizados. Aproximadamente metade dos pacientes de cada grupo de tratamento completou o estudo (53,6% dos pacientes randomizados com CBZ e 54% dos pacientes randomizados com LEV). A distribuição pela categoria do tipo de crise foi similar em ambos os grupos de tratamento, com cerca de 86,7% dos pacientes classificados por terem experimentado crises focais/parciais com clara origem focal. A maioria dos pacientes permaneceram no nível de dose 1 (81,7% dos pacientes randomizados com CBZ e 73,4% dos pacientes randomizados com LEV na população PP - população avaliada por protocolo).
Um desfecho primário definido prospectivamente foi a proporção de pacientes da população PP há 6 meses livres de crises na última dose avaliada.
Cento e setenta e três (73%) dos pacientes PP no braço LEV estavam livres de crises por pelo menos 6 meses na última dose avaliada, comparado com 171 pacientes (72,8%) no braço CBZ. A diferença absoluta ajustada entre LEV e CBZ (IC 95% bicaudal) obtida de um modelo de regressão logística incluindo um fator para uma categoria de crise como a última avaliada (IA/IB/IC versus IC/IIE) foi igual a 0,2% (-7,8%; 8,2%). O limite inferior do intervalo de confiança (-7,8%) foi acima do limite de não inferioridade determinado pelo protocolo (-15%) para esta análise de eficácia primária, e portanto, LEV pode ser considerado não inferior a CBZ na proporção de sujeitos livres de crises por pelo menos 6 meses na primeira dose avaliada na população PP. Considerando outro desfecho clinicamente significante, 56,6% e 58,5% dos pacientes de LEV e CBZ, respectivamente, ficaram livres de crises por 1 ano.
- Terapia adjuvante:
A eficácia de levetiracetam como terapia adjuvante (adicionada a outras drogas antiepilépticas) em adultos foi estabelecida em três estudos clínicos multicêntricos, randomizados, duplo-cego, placebo controlado, em pacientes que tiveram crises focais/parciais refratárias com ou sem generalização secundária. A formulação em comprimidos foi utilizada em todos esses estudos. Nesses estudos, 904 pacientes foram randomizados com placebo, 1000 mg, 2000 mg ou 3000 mg/dia. Os pacientes inscritos no Estudo 1 ou no Estudo 2 tiveram crises focais/parciais refratárias por pelo menos 2 anos e usaram 2 ou mais fármacos antiepilépticos (FAEs) clássicos. Os pacientes incluídos no Estudo 3 tiveram crises refratárias por pelo menos 1 ano e utilizaram pelo menos um FAE clássico. No período do estudo, pacientes estavam utilizando um regime de dose estável de pelo menos um e poderiam utilizar no máximo dois FAEs. Durante o período basal, pacientes tiveram pelo menos duas crises focais/parciais durante cada período de 4 semanas.
Estudo 1:
O Estudo 1 foi um estudo duplo-cego, placebo controlado, grupo paralelo conduzido em 41 centros nos Estados Unidos comparando levetiracetam 1000 mg/dia (N = 98), levetiracetam 3000 mg/dia (N = 101) e placebo (N = 95) administrado em doses igualmente divididas duas vezes ao dia.
Após um período basal prospectivo de 12 semanas, pacientes foram randomizados para um dos três grupos de tratamento descritos acima. Um tratamento de 18 semanas consistiu em um período de 6 semanas seguidos por um período de avaliação de dose fixa de 12 semanas, durante as quais regimes de FAEs se mantiveram constantes. A medida primária de eficácia foi uma comparação entre grupos da redução percentual da frequência das crises focais/parciais semanais em comparação ao placebo ao longo de todo o período de avaliação (considerando as últimas 2 semanas de titulação + 12 semanas do período de avaliação). Variáveis secundárias de resultado incluíram a taxa de respondedores (incidência de pacientes com uma redução maior ou igual a 50% a partir do basal na frequência de crises focais/parciais). Os resultados da análise do Estudo 1 estão disponíveis na Tabela 1.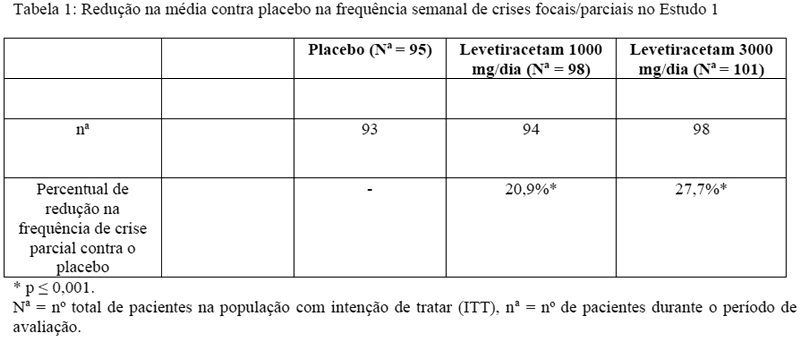
A porcentagem de pacientes (eixo y) que alcançaram uma redução maior ou igual a 50% nas taxas de crises semanais a partir do basal na frequência de crises focais/parciais ao longo do período de avaliação (considerando as 2 últimas semanas da titulação + 12 semanas do período de avaliação) nos 3 grupos de estudo (eixo x) está representada na Figura 1: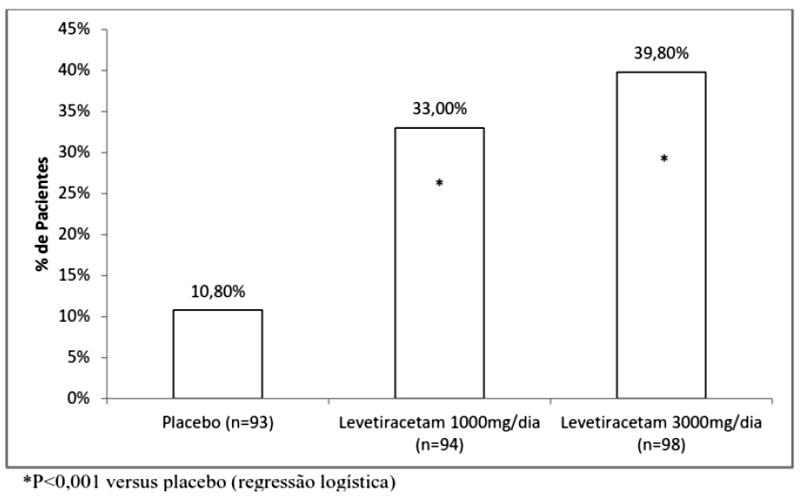
Estudo 2:
O Estudo 2 foi um estudo duplo-cego, placebo controlado, cruzado, conduzido em 62 centros na Europa comparando levetiracetam 1000 mg/dia (N=106), levetiracetam 2000 mg/dia (N=106) e placebo (N=112) administrado em doses divididas igualmente duas vezes ao dia.
O primeiro período do estudo (Período A) foi desenhado para ser analisado como um estudo de grupo paralelo. Após um período de base prospectivo de até 12 semanas, os pacientes foram randomizados em um dos três grupos de tratamento descritos acima. Um período de tratamento de 16 semanas consistiu em um período de titulação de 4 semanas, seguidos por um período de avaliação de dose fixa de 12 semanas, durante as quais regimes concomitantes de FAEs foram mantidos constantes. O desfecho primário de eficácia foi uma comparação entre a redução percentual na frequência de crises focais/parciais semanais em relação ao placebo durante o período de avaliação. As variáveis secundárias dos resultados incluíram a taxa de resposta (incidência de pacientes com uma redução maior ou igual a 50% a partir do basal na frequência de crise parcial). Os resultados das análises do Período A estão disponíveis na Tabela 2.
A porcentagem de pacientes (eixo y) que alcançaram uma redução maior ou igual a 50% nas taxas de crises semanais a partir do basal na frequência de crises focais/parciais ao longo do período de tratamento randomizado (titulação + período de avaliação) nos 3 grupos de estudo (eixo x) está representada na Figura 2: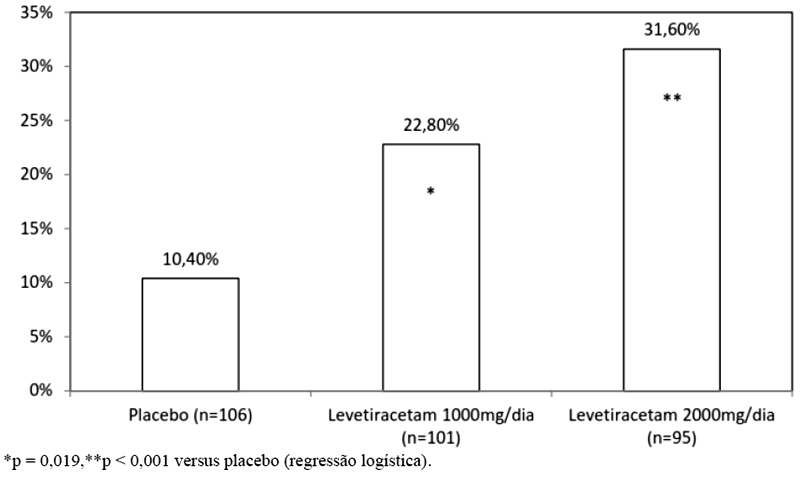
- Estudo 3
O Estudo 3 foi um estudo duplo-cego, placebo controlado, grupo paralelo, conduzido em 47 centros na Europa comparando levetiracetam 3000 mg/dia (N = 181) e placebo (N = 105) administrados em pacientes com crises focais/parciais refratárias, com ou sem generalização secundária, recebendo somente um FAE concomitante. A droga em estudo foi administrada em duas doses divididas.
Após um período de base de 12 semanas, os pacientes foram randomizados em um dos dois grupos de tratamento descritos acima. O período de tratamento de 16 semanas consistiu em um período de titulação de 4 semanas, seguido por um período de avaliação de 12 semanas com dose fixa de levetiracetam administrado como terapia adjuvante (add-on) e um período de 2 semanas para seleção de respondedores, durante o qual doses concomitantes de FAEs foram mantidas constantes. O desfecho primário de eficácia foi a comparação entre grupos da redução percentual da frequência de crises semanais em relação ao placebo ao longo de todo o período randomizado de avaliação add-on (considerando 12 semanas de avaliação com dose fixa de levetiracetam administrado como terapia adjuvante + 2 semanas do período de seleção de respondedores). As variáveis de desfecho secundário incluíram a taxa de respondedores (incidência de pacientes com uma redução maior ou igual a 50% a partir do basal na frequência de crises focais/parciais). A Tabela 3 possui os resultados da análise do Estudo 3.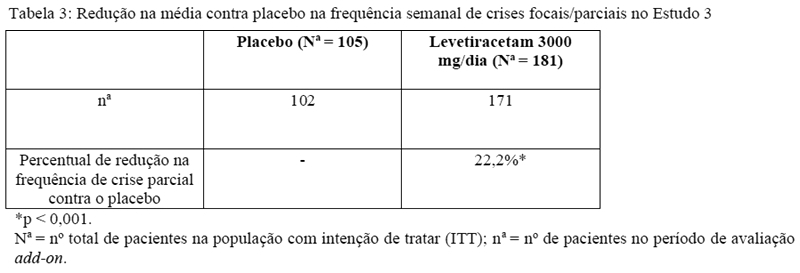
A porcentagem de pacientes (eixo y) que alcançaram uma redução maior ou igual a 50% nas taxas de crises semanais a partir do basal na frequência de crises focais/parciais ao longo do período de avaliação add-on (considerando 12 semanas de avaliação com dose fixa de levetiracetam administrado como terapia adjuvante + 2 semanas do período de seleção de respondedores) nos 2 grupos de estudo (eixo x) está representada na Figura 3: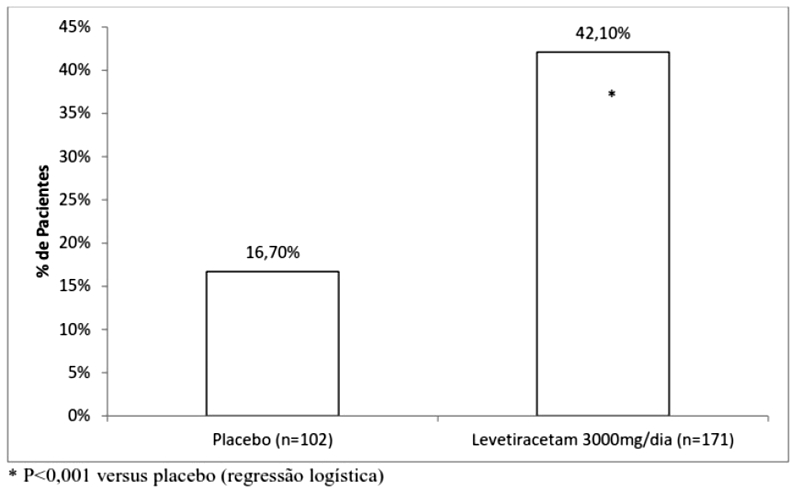
Em uma análise agrupada destes três estudos, a porcentagem de pacientes que alcançaram 50% ou mais de redução a partir do basal na frequência de crises focais/parciais por semana em uma dose estável (12/14 semanas) foi de 27,7%, 31,6% e 41,3% para pacientes com 1000, 2000 ou 3000 mg de levetiracetam respectivamente, e 12,6% para pacientes recebendo placebo.
Crises focais/parciais em pacientes pediátricos com epilepsia
- População pediátrica (4 a 16 anos de idade)
A eficácia de levetiracetam como terapia adjuvante (juntamente com outras drogas antiepilépticas) em pacientes pediátricos foi estabelecida em um estudo multicêntrico, randomizado, duplo-cego, placebo controlado, conduzido em 60 centros na América do Norte, em crianças de 4 a 16 anos de idade com crises focais/parciais não controladas por fármacos antiepilépticos (FAEs) padrão. Pacientes elegíveis com uma dose estável de 1-2 FAEs, que ainda vivenciaram pelo menos 4 crises focais/parciais durante as 4 semanas antes da triagem, assim como pelo menos 4 crises focais/parciais em cada um dos dois períodos de 4 semanas do período basal, foram randomizados para receber levetiracetam ou placebo. A população inscrita incluiu 198 pacientes (levetiracetam = 101, placebo = 97) com crises refratárias focais/parciais, com ou sem generalização secundária. O estudo consistiu em um período basal de 8 semanas e um período de titulação de 4 semanas seguido por um período de avaliação de 10 semanas. O doseamento teve início com uma dose de 20 mg/kg/dia em duas doses divididas. Durante o período de tratamento, as doses de levetiracetam foram ajustadas em incrementos de 20 mg/kg/dia, com intervalos de 2 semanas para a dose alvo de 60 mg/kg/dia. O desfecho primário de eficácia foi a comparação entre grupos do percentual de redução na frequência de crises focais/parciais semanais em relação ao placebo durante todo o período randomizado de 14 semanas (titulação + período de avaliação). As variáveis de desfecho secundário incluíram a taxa de respondedores (incidência de pacientes com redução maior ou igual a 50% a partir do basal na frequência de crises focais/parciais por semana). Na Tabela 4 estão disponibilizados os resultados deste estudo:
A porcentagem de pacientes (eixo y) que alcançaram uma redução maior ou igual a 50% nas taxas de crises semanais a partir do basal na frequência de crises focais/parciais ao longo de todo o período de tratamento randomizado (titulação + período de avaliação) dentro dos dois grupos de tratamento (eixo x) é apresentada na Figura 4.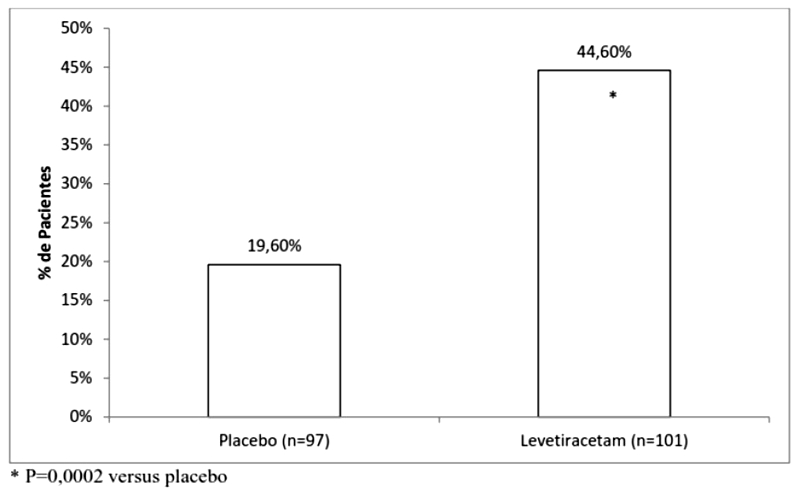
Com a continuidade do tratamento de longo prazo, 11,4% dos pacientes ficaram livres de crises por pelo menos 6 meses e 7,2% ficaram livres de crises por pelo menos 1 ano.
Crises mioclônicas em pacientes ≥ 12 anos de idade
A eficácia de levetiracetam como terapia adjuvante (juntamente com outras drogas antiepilépticas) em pacientes de 12 anos de idade ou mais com epilepsia mioclônica juvenil foi estabelecida em um estudo multicêntrico, randomizado, duplo-cego, placebo controlado, conduzido em 37 centros de 14 países. Pacientes elegíveis com uma dose estável de 1 FAE que vivenciaram uma ou mais crises mioclônicas por dia por pelo menos 8 dias durante o período basal de 8 semanas foram randomizados tanto para levetiracetam quanto para placebo. A população inscrita incluiu 120 pacientes (levetiracetam = 60, placebo = 60) com epilepsia idiopática generalizada, que incluiu epilepsia mioclônica juvenil, epilepsia tipo ausência juvenil ou epilepsia com crises tônico-clônicas generalizadas ao despertar. A maioria era de pacientes com epilepsia mioclônica juvenil. Pacientes foram titulados por 4 semanas com uma dose alvo de 3000 mg/dia e tratados com uma dose estável de 3000 mg/dia por 12 semanas (período de avaliação). A droga de estudo foi administrada em duas doses. O desfecho primário de eficácia foi a proporção de pacientes com uma redução de pelo menos 50% no número de dias por semana com uma ou mais crises mioclônicas durante o período de tratamento (titulação + período de avaliação) quando comparado com o basal. Variáveis de desfecho secundário incluíram ausência de crise (crises mioclônicas) e taxa de resposta na frequência de crise mioclônica por semana durante o período de tratamento. A Tabela 5 apresenta os resultados deste estudo para o desfecho primário de eficácia.
Com a continuidade do tratamento a longo prazo, 28,6% dos pacientes ficaram livres das crises mioclônicas por pelo menos 6 meses e 21% ficaram livres das crises mioclônicas por pelo menos 1 ano.
Crises tônico-clônicas primárias generalizadas em pacientes ≥ 6 anos de idade
A eficácia de levetiracetam como terapia adjuvante (juntamente com outras drogas antiepilépticas) em pacientes de 6 anos de idade ou mais com epilepsia idiopática generalizada com crises tônico-clônicas primárias generalizadas (PGTC) foi estabelecida em um estudo multicêntrico, randomizado, duplo-cego, placebo controlado, conduzido em 50 centros de 8 países. Pacientes elegíveis com uma dose estável de 1 ou 2 FAEs que vivenciaram pelo menos 3 crises PGTC durante o período basal combinado (pelo menos uma convulsão PGTC durante 4 semanas antes do período basal prospectivo e pelo menos uma crise PGTC durante as 4 semanas do período basal prospectivo) foram randomizados para levetiracetam ou para placebo. O período basal combinado de 8 semanas é referido como "linha basal" no restante desta seção. A população incluída foi de 164 pacientes (levetiracetam = 80, placebo = 84) com epilepsia idiopática generalizada (predominantemente epilepsia mioclônica juvenil, epilepsia ausência juvenil, epilepsia ausência infantil ou crises generalizadas tônico-clônicas ao despertar). Cada uma dessas síndromes de epilepsia idiopática generalizada foi bem representada nesta população de pacientes.
Os pacientes foram titulados por 4 semanas com uma dose alvo de 3000 mg/dia para adultos ou uma dose alvo pediátrica de 60 mg/kg/dia e tratados com uma dose estável de 3000 mg/dia (ou 60 mg/kg/dia para crianças) por 20 semanas (período de avaliação). A droga de estudo foi administrada em duas doses igualmente divididas/dia.
O desfecho primário de eficácia foi a redução percentual a partir do basal na frequência das crises PGTC semanais para os grupos de tratamento com levetiracetam e placebo durante o período de tratamento (titulação + período de avaliação). Houve uma diminuição estatisticamente significativa a partir do basal da frequência de PGTC nos pacientes tratados com levetiracetam em comparação aos pacientes tratados com placebo. A significância estatística versus placebo indica um valor p < 0,05.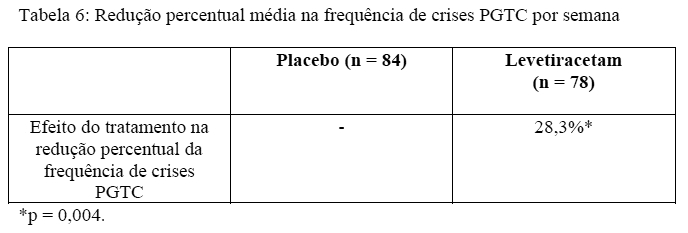
A porcentagem de pacientes (eixo y) que alcançaram uma redução maior ou igual a 50% nas taxas de crises semanais a partir do basal na frequência de crises PGTC durante todo o período de tratamento randomizado (titulação + período de avaliação) nos dois grupos de tratamento (eixo x) é apresentada na Figura 6.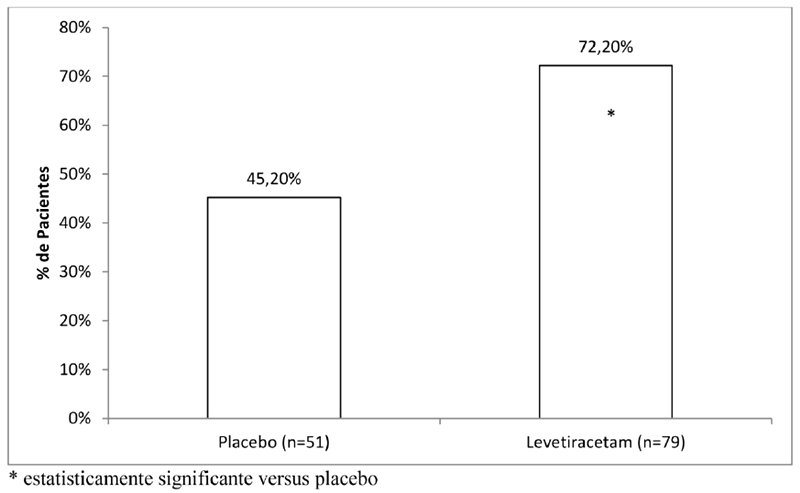
Com a continuidade do tratamento a longo prazo, 47,4% dos pacientes ficaram livres das crises tônico-clônicas por pelo menos 6 meses e 31,5% ficaram livres das crises tônico-clônicas por pelo menos 1 ano.
Referências:
- Clinical study report N01061: A multicenter, double-blind, randomized, parallel-group, positive controlled trial comparing the efficacy and safety of levetiracetam (1000 to 3000 mg/day oral b.i.d.) to carbamazepine (400 to 1200 mg/day oral b.i.d.), used as monotherapy for up to a maximum of 121 weeks in subjects (≥ 16 years) newly or recently diagnosed as suffering from epilepsy, and experiencing partial or generalized tonic-clonic seizures.
- Clinical study report N132: Evaluation of the efficacy and tolerability of ucb L059 (500 and 1500 mg b.i.d., tablets) add-on treatment in epileptic patients with partial onset seizures: a 38- week double-blind placebo-controlled parallel multicenter trial.
- Clinical study report N051: Evaluation of the efficacy and tolerability of ucb L059 (500 and 1000 mg b.i.d., tablets) add-on treatment in refractory epileptic patients with partial onset seizures: a 32-week double-blind placebo-controlled crossover multicenter trial.
- Clinical study report N138: Evaluation of the efficacy and tolerability of ucb L059 (1500 mg b.i.d., 500 mg tablets) monotherapy in epileptic patients with complex partial onset seizures, having experienced improved seizure control under add-on treatment.
- Clinical study report N159: Evaluation of The Efficacy and Tolerability of Levetiracetam Add-On Treatment in Refractory Pediatric Patients With Partial Onset Seizures: A 28Week Double-Blind, Placebo-Controlled Multi-Center Trial (4 years to < 16 years)
- Clinical study report N166: A double-blind, multicenter, randomized, placebo-controlled study to evaluate the efficacy and safety of levetiracetam (LEV) (oral tablets of 500 mg b.i.d.), at a dose of 3000 mg/day as adjunctive treatment in adolescents (≥ 12 years) and adults (≤ 65 years) suffering from idiopathic generalized epilepsy with myoclonic seizures.
- Clinical study report N01057: A double-blind, multicenter, randomized, placebo-controlled study to evaluate the efficacy and safety of adjunctive treatment with 3000 mg/day (pediatric target dose of 60 mg/kg/day) oral levetiracetam (LEV) (166, 250, and 500 mg tablets), in adult and pediatric subjects (4-65 years) suffering from idiopathic generalized epilepsy with primary generalized tonic-clonic (PGTC) seizures.
3. CARACTERÍSTICAS FARMACOLÓGICAS
PROPRIEDADES FARMACODINÂMICAS
A substância ativa, levetiracetam, é um derivado da pirrolidona (enantiômero-S de a-etil-2-oxo1-pirrolidina acetamida), quimicamente não relacionada com substâncias ativas antiepilépticas existentes.
Mecanismo de ação:
O mecanismo de ação do levetiracetam ainda não é elucidado completamente, mas parece ser diferente dos mecanismos antiepilépticos já existentes. Experiências in vitro e in vivo sugerem que o levetiracetam não altera as características básicas da célula nem a neurotransmissão normal. Estudos in vitro mostram que o levetiracetam afeta os níveis de Ca2+ intraneuronais, pela inibição parcial das correntes de Ca2+ das reservas intraneuronais. Adicionalmente, reverte parcialmente as reduções nas correntes de entrada do GABA e da glicina, induzidas pelo zinco e pelas ß-carbolinas. Além disto, em estudos in vitro demonstrou-se que o levetiracetam se liga a um local específico no tecido cerebral dos roedores. Este local de ligação é a proteína 2A da vesícula sináptica, que se pensa estar envolvida na fusão das vesículas e na exocitose dos neurotransmissores. O levetiracetam e análogos relacionados mostram uma ordem de grandeza de afinidade para a ligação com proteína 2A da vesícula sináptica, que se correlaciona com a potência da sua proteção anticonvulsivante, no modelo audiogênico de epilepsia em camundongo. Este resultado sugere que a interação entre o levetiracetam e a proteína 2A da vesícula sináptica parece contribuir para o mecanismo de ação antiepiléptica do fármaco.
Efeitos farmacodinâmicos:
O levetiracetam induz proteção contra a crise em um grande número de modelos animais de crises focais/parciais e primárias generalizadas sem apresentar um efeito pró-convulsivante. O metabólito primário é inativo. No homem, uma atividade em ambas as condições de epilepsia parcial e generalizada (descarga epileptiforme/resposta fotoparoxística) confirmou o perfil farmacológico pré-clínico de largo espectro.
PROPRIEDADES FARMACOCINÉTICAS
O levetiracetam é um composto altamente solúvel e permeável. O perfil farmacocinético é linear com uma baixa variabilidade intra e interindividual. Não há alteração da depuração após administração repetida. Não há evidência de qualquer variabilidade relevante relacionada com o sexo, raça ou ritmo circadiano. O perfil farmacocinético é comparável em voluntários saudáveis e em pacientes com epilepsia.
Devido à sua absorção completa e linear, os níveis plasmáticos podem ser deduzidos a partir da dose oral de levetiracetam expressa em mg/kg de peso corporal. Deste modo, não é necessária a monitorização dos níveis plasmáticos de levetiracetam.
Foi demonstrada uma correlação significativa entre as concentrações na saliva e no plasma, em adultos e crianças (a relação entre concentrações na saliva/plasma variou de 1 a 1,7 para a formulação dos comprimidos orais e 4 horas após administração para a formulação da solução oral).
- Absorção:
O levetiracetam é rapidamente absorvido após administração por via oral. A biodisponibilidade oral absoluta é próxima a 100%.
Os picos das concentrações plasmáticas (Cmáx) são atingidos 1,3 horas após a administração. O estado de equilíbrio é atingido 2 dias após um esquema de administração de duas vezes por dia.
Os picos das concentrações (Cmáx) são habitualmente de 31 e 43 mg/mL, após uma dose única de 1000 mg e de uma dose repetida de 1000 mg duas vezes por dia, respectivamente.
A extensão de absorção é independente da dose e não é alterada pelos alimentos.
- Distribuição:
Não existem dados disponíveis sobre a distribuição nos tecidos em humanos.
Nem o levetiracetam, nem o metabólito primário se ligam significativamente às proteínas plasmáticas ( < 10%).
O volume de distribuição do levetiracetam é aproximadamente de 0,5 a 0,7 L/kg, um valor próximo do volume de água corporal total.
- Biotransformação:
O levetiracetam não é extensivamente metabolizado nos humanos. A principal via metabólica (24% da dose) é uma hidrólise enzimática do grupo acetamida. A produção do metabólito primário, ucb L057, não é suportado pelas isoformas do citocromo hepático P450. A hidrólise do grupo acetamida foi determinável em um vasto número de tecidos incluindo as células sanguíneas.
O metabólito ucb L057 é farmacologicamente inativo.
Dois metabólitos menores também foram identificados. Um deles foi obtido por hidroxilação do anel pirrolidona (1,6% da dose) e o outro pela abertura do anel pirrolidona (0,9% da dose).
Outros componentes não identificados foram responsáveis por apenas 0,6% da dose.
Não foi evidenciada qualquer interconversão enantiomérica in vivo para o levetiracetam ou para o seu metabólito primário.
O levetiracetam e seu metabólito primário tem mostrado, in vitro, não inibir as isoformas principais do citocromo hepático humano P450 (CYP3A4, 2A6, 2C9, 2D6, 2C19, 2D6, 2E1 e 1A2), a glucuronil transferase (UGT1A1 e UGT1A6) e as atividades da epóxido-hidroxilase.
Além disso, o levetiracetam não afeta a glucuronidação in vitro do ácido valproico.
Em hepatócitos humanos em cultura, o levetiracetam teve efeito mínimo ou ausência de efeito sobre a conjugação do etinilestradiol ou CYP1A1/2. O levetiracetam provocou indução moderada sobre CYP2B6 e CYP3A4 em altas concentrações (680 mg/mL), porém, em concentrações aproximadas do Cmáx seguindo-se duas doses diárias de 1500 mg, os efeitos não foram considerados biologicamente relevantes. Deste modo, a interação de levetiracetam com outras substâncias, ou vice-versa, é pouco provável.
- Eliminação:
A meia-vida plasmática em adultos foi 7 ± 1 hora e não se alterou com a dose, a via de administração ou com a administração repetida. A depuração corporal total média foi 0,96 mL/min/kg.
A principal via de excreção é a via urinária, sendo responsável por 95% da dose (aproximadamente 93% da dose foi excretada no intervalo de 48 horas). A excreção via fecal foi responsável por apenas 0,3% da dose.
A excreção urinária cumulativa do levetiracetam e do seu metabólito primário foi responsável por 66% e 24% da dose, respectivamente durante as primeiras 48 horas.
A depuração renal do levetiracetam e do ubc L057 é de 0,6 e 4,2 mL/min/kg, respectivamente, indicando que o levetiracetam é excretado por filtração glomerular com subsequente reabsorção tubular e que o metabólito primário é igualmente excretado por secreção tubular ativa, em adição à filtração glomerular. A eliminação do levetiracetam está correlacionada com a depuração da creatinina.
- Idosos:
Nos idosos, a meia-vida é aumentada em cerca de 40% (10 a 11 horas). Isto está relacionado com a diminuição da função renal nestes indivíduos (vide item 8. POSOLOGIA E MODO DE USAR).
- População pediátrica:
- Crianças (4 aos 12 anos)
Após uma administração oral de dose única (20 mg/dia) a crianças epiléticas, a meia-vida do levetiracetam foi de 6 horas. A depuração corporal aparente foi de 1,43 mL/min/kg.
Após uma administração de doses orais repetidas (20 a 60 mg/dia) a crianças epiléticas (4 a 12 anos), o levetiracetam foi rapidamente absorvido. O pico da concentração plasmática foi observado 0,5 a 1 hora após a administração. Foram observados aumentos lineares e proporcionais à dose para o pico da concentração plasmática e para a área sob a curva. A meia-vida de eliminação foi de, aproximadamente, 5 horas. A depuração corporal aparente foi de 1,1 mL/min/kg.
Na análise farmacocinética populacional efetuada em pacientes com idades entre 1 mês e 16 anos após administração oral, o peso corporal teve uma correlação significativa com a depuração aparente (a depuração aumentou com o aumento do peso corporal) e com o volume de distribuição aparente. A idade também teve influência em ambos os parâmetros. Este efeito foi mais pronunciado nas crianças mais novas, diminuindo com o aumento da idade, até se tornar negligenciável por volta dos 4 anos de idade.
Nas análises farmacocinéticas populacionais verificou-se um aumento de cerca de 20% na depuração aparente do levetiracetam quando este foi coadministrado com fármaco antiepiléptico (FAE) indutor enzimático.
- Comprometimento renal:
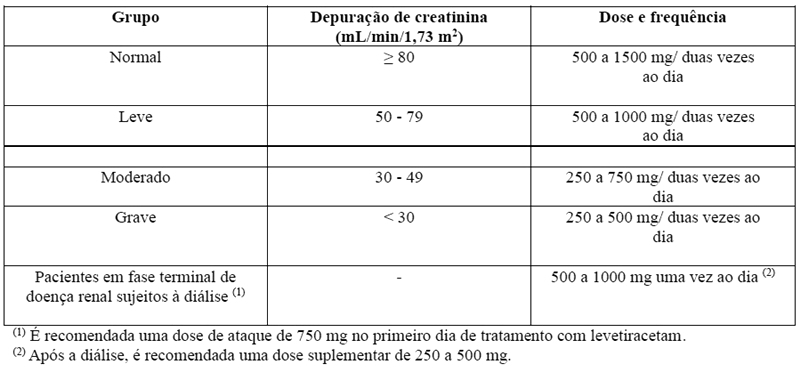
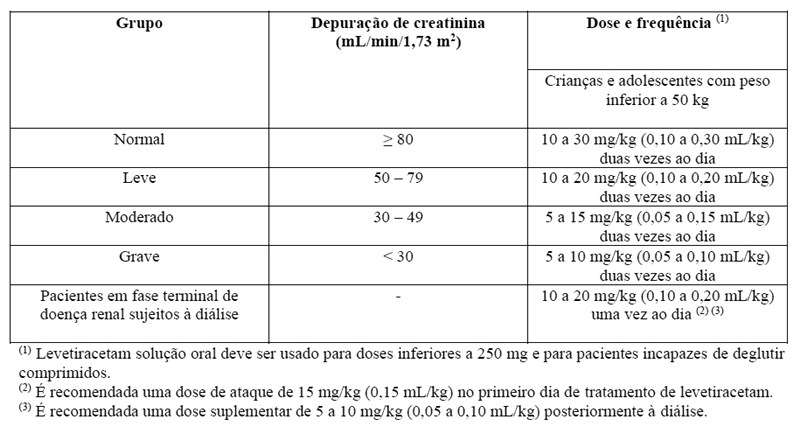
A depuração corporal aparente de levetiracetam e do seu metabólito primário está correlacionada com a depuração da creatinina. Recomenda-se, além disso, o ajuste da dose diária de manutenção de levetiracetam, com base na depuração de creatinina em pacientes com comprometimento renal moderado e grave (vide item 8. POSOLOGIA E MODO DE USAR).
Nos indivíduos adultos em fase anúrica terminal, a meia-vida foi aproximadamente 25 e 3,1 horas, durante períodos inter e intradiálise, respectivamente.
A remoção fracional do levetiracetam foi de 51%, durante uma sessão comum de diálise de 4 horas.
- Comprometimento hepático:
Em indivíduos com comprometimento hepático leve (Child-Pugh A) e moderado (Child-Pugh B), a farmacocinética de levetiracetam permaneceu inalterada. Na maioria dos indivíduos com comprometimento hepático grave (Child-Pugh C), a depuração corpórea total foi de 50% comparado a pacientes normais, mas a diminuição da depuração renal foi responsável pela maior parte da diminuição (vide item 8. POSOLOGIA E MODO DE USAR).
4. CONTRAINDICAÇÕES
Hipersensibilidade ao princípio ativo ou a outros derivados da pirrolidona ou a qualquer um dos excipientes.
Categoria de risco na gravidez: C.
Este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica ou do cirurgião-dentista.
5. ADVERTÊNCIAS E PRECAUÇÕES
- Descontinuação
De acordo com a prática clínica atual, se levetiracetam tiver que ser descontinuado, recomenda-se que a sua descontinuação seja efetuada de forma gradual (exemplo: nos adultos e adolescentes com peso superior a 50 kg: reduções de 500 mg duas vezes ao dia, a cada duas a quatro semanas; nas crianças com mais de 6 meses de idade e adolescentes com menos de 50 kg de peso, a diminuição da dose não deve exceder 10 mg/kg duas vezes ao dia, a cada duas semanas).
- Contagem de células sanguíneas
Casos de diminuição na contagem de células sanguíneas (neutropenia, agranulocitose, leucopenia, trombocitopenia e pancitopenia) foram descritos em associação à administração de levetiracetam. Contagens completas de células sanguíneas são recomendadas em pacientes apresentando fraqueza, pirexia (febre), infecções recorrentes ou distúrbios de coagulação (vide item 9. REAÇÕES ADVERSAS).
- Insuficiência renal
A administração de levetiracetam em pacientes com comprometimento renal poderá necessitar de um ajuste da dose. Em pacientes com comprometimento grave da função hepática, recomenda-se a avaliação da função renal antes de selecionar a dose (vide item 8. POSOLOGIA E MODO DE USAR).
- Suicídio
Foram notificados suicídio, tentativa de suicídio e ideias e comportamento suicidas em pacientes tratados com levetiracetam.
Os pacientes devem ser aconselhados a contatar o médico assim que surjam sinais de depressão e/ou ideias e comportamento suicidas.
- Transtornos psiquiátricos e alterações no comportamento
Levetiracetam pode causar alterações no comportamento (por exemplo, agressividade, agitação, raiva, ansiedade, apatia, depressão, hostilidade e irritabilidade) e sintomas psicóticos. Os pacientes tratados com levetiracetam devem ser monitorizados quanto a sinais e sintomas psiquiátricos.
-Agravamento de crises epilépticas
Pode ser observada uma reação paradoxal do agravamento de crises epilépticas, especialmente ao iniciar o tratamento ou no aumento da dose.
Fertilidade, gravidez e lactação
- Gravidez
O levetiracetam não deve ser usado durante a gravidez a menos que seja clinicamente necessário.
Dados de pós-comercialização de alguns registros de gravidez documentaram os resultados em mais de 1000 mulheres expostas à monoterapia com levetiracetam durante o primeiro trimestre de gravidez. No geral, estes dados não sugerem um aumento substancial no risco de malformações importantes, embora este não possa ser completamente excluído. O tratamento com múltiplos medicamentos antiepilépticos está associado a um risco maior de malformações em comparação à monoterapia, e portanto, esta deve ser considerada.
Estudos em animais revelaram toxicidade reprodutiva (vide Dados de segurança pré-clínica). Tal como acontece com outros medicamentos antiepilépticos, as alterações fisiológicas durante a gravidez podem afetar a concentração de levetiracetam. Houve relatos de diminuição na concentração de levetiracetam durante a gravidez.
Esta diminuição é mais pronunciada durante o terceiro trimestre (até 60% da concentração basal antes da gravidez). A descontinuação dos tratamentos antiepilépticos pode resultar na exacerbação da doença, o que pode ser perigoso para a mãe e para o feto. A gravidez de mulheres em tratamento com levetiracetam deve ser monitorada.
Categoria de risco na gravidez: C.
Este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica ou do cirurgião-dentista.
- Lactação
O levetiracetam é excretado no leite humano materno. Portanto, a amamentação não é recomendada. No entanto, se o tratamento com levetiracetam for necessário durante a amamentação, o benefício/risco do tratamento deve ser avaliado tendo em consideração a importância da amamentação.
Efeitos na habilidade de dirigir ou operar máquinas
Não foram estudados os efeitos sobre a capacidade de conduzir veículos e utilizar máquinas.
Devido a possíveis sensibilidades individuais diferentes, alguns pacientes poderão apresentar sonolência ou outros sintomas relacionados com o sistema nervoso central, especialmente no início do tratamento ou após um aumento da dose. Assim sendo, recomenda-se precaução nos pacientes que executam tarefas especializadas, por exemplo, condução de veículos ou utilização de máquinas.
Dados de segurança pré-clínica
Os dados pré-clínicos não revelam riscos especiais em humanos, segundo estudos convencionais de farmacologia de segurança, genotoxicidade e carcinogenicidade.
Efeitos adversos não observados nos estudos clínicos, mas verificados em ratos e em menor grau em camundongos, em níveis de exposição semelhantes aos níveis de exposição em humanos e com possível relevância para o uso clínico foram: alterações hepáticas indicando uma resposta adaptativa, tais como um aumento de peso e hipertrofia centrolobular, infiltração lipídica e aumento das enzimas hepáticas no plasma.
Carcinogênese
O levetiracetam foi administrado em ratos durante 104 semanas em doses de 50, 300 e 1800 mg/kg/dia. A dose mais alta corresponde a 6 vezes a dose máxima diária recomendada para humanos (3000 mg) em mg/m2. Esta dose também levou a uma exposição sistêmica (AUC) de aproximadamente 6 vezes a obtida com a dose diária máxima em humanos. Não houve evidência de carcinogenicidade. Dois estudos foram conduzidos em camundongos. Em um estudo, os camundongos receberam levetiracetam na dieta por 80 semanas em níveis de dose de 60, 240 e 960 mg/kg/dia (a dose mais alta é equivalente a 2 vezes a dose máxima diária em mg/m2 ou em termos de exposição). No segundo estudo, os camundongos receberam levetiracetam através de sonda oral durante 2 anos em níveis de dose de 1000, 2000 e 4000 mg/kg/dia. Devido à baixa sobrevida com a dose de 4000 mg/kg/dia, neste estudo, a dose mais alta foi reduzida para 3000 mg/kg/dia (equivalente a 12 vezes a dose máxima diária recomendada em humanos).
Nenhum dos estudos mostrou evidência de carcinogenicidade.
Foram efetuados dois estudos de desenvolvimento embriofetal (EFD) em ratos com doses de 400, 1200 e 3600 mg/kg/dia. Com a dose de 3600 mg/kg/dia observou-se, em apenas um dos dois estudos EFD, uma ligeira diminuição no peso fetal associada a um aumento marginal de anomalias menores/alterações esqueléticas. Não foram observados efeitos sobre a mortalidade embrionária e não houve aumento da incidência de malformações. O NOAEL (nível de efeito adverso não observável) foi de 3600 mg/kg/dia para ratas grávidas (12 vezes a dose máxima diária recomendada para humanos em mg/m2) e 1200 mg/kg/dia para fetos.
Foram efetuados quatro estudos de desenvolvimento embriofetal em coelhos abrangendo as doses de 200, 600, 800, 1200 e 1800 mg/kg/dia. A dose de 1800 mg/kg/dia induziu uma toxicidade maternal marcada e uma diminuição no peso fetal associada ao aumento de incidência de fetos com anomalias cardiovasculares/esqueléticas. O NOAEL foi < 200 mg/kg/dia para as mães e 200 mg/kg/dia para os fetos (igual à dose máxima diária recomendada para humanos, considerando mg/m2).
Foi efetuado um estudo de desenvolvimento peri e pós-natal em ratos com doses de levetiracetam de 70, 350 e 1800 mg/kg/dia. O NOAEL foi ≥ 1800 mg/kg/dia para as fêmeas F0, e para a sobrevivência, crescimento e desenvolvimento da ninhada F1 até o desmame (6 vezes a dose máxima diária recomendada para humanos, considerando mg/m2).
Estudos animais realizados em ratos e cães recém-nascidos e jovens demonstraram que não ocorreram efeitos adversos sobre nenhum dos desfechos padronizados para avaliação do desenvolvimento e de maturação, com doses de até 1800 mg/kg/dia (6-17 vezes a dose máxima diária recomendada para humanos, considerando mg/m2).
Atenção diabéticos: contém açúcar.
6. INTERAÇÕES MEDICAMENTOSAS
- Medicamentos antiepilépticos
Dados indicam que levetiracetam não influencia as concentrações séricas de medicamentos antiepilépticos existentes (fenitoína, carbamazepina, ácido valproico, fenobarbital, lamotrigina, gabapentina e primidona) e que estes medicamentos antiepilépticos não influenciam a farmacocinética de levetiracetam.
A depuração de levetiracetam foi 22% mais alta em crianças utilizando FAEs indutores enzimáticos comparado com crianças que não estavam utilizando FAEs indutores enzimáticos. O ajuste de dose não é recomendado. O levetiracetam não teve qualquer efeito nas concentrações plasmáticas de carbamazepina, valproato, topiramato ou lamotrigina.
- Probenecida
A probenecida (500 mg quatro vezes ao dia),