AFINITORTM
NOVARTIS
everolimo
Antineoplásico.
Apresentações.
Comprimidos
Embalagens com 30 comprimidos de 2,5 mg, 5 mg ou 10 mg.
VIA ORAL
USO ADULTO E PEDIÁTRICO ACIMA DE 3 ANOS
Composição.
Cada comprimido de AfinitorTM 2,5 mg, 5 mg e 10 mg contém respectivamente 2,5 mg, 5 mg e 10 mg de everolimo.
Excipientes: lactose anidra, crospovidona, hipromelose, lactose monoidratada, estearato de magnésio e butilidroxitolueno.
Indicações.
AfinitorTM é indicado para o tratamento de:
- Tumores neuroendócrinos avançados (NET) localizados no estômago e intestino, pulmão ou pâncreas.
- Câncer avançado do(s) rim(ns) (Carcinoma avançado de Células Renais (CCR)) cuja doença tenha progredido durante ou após o tratamento com VEGFR - TKI, quimioterápicos ou imunoterápicos.
- Astrocitoma subependimário de células gigantes (SEGA, um tumor cerebral específico) associado à esclerose tuberosa (TS).
Resultados de eficácia.
Tumores neuroendócrinos avançados de origem gastrintestinal, pulmonar ou pancreática
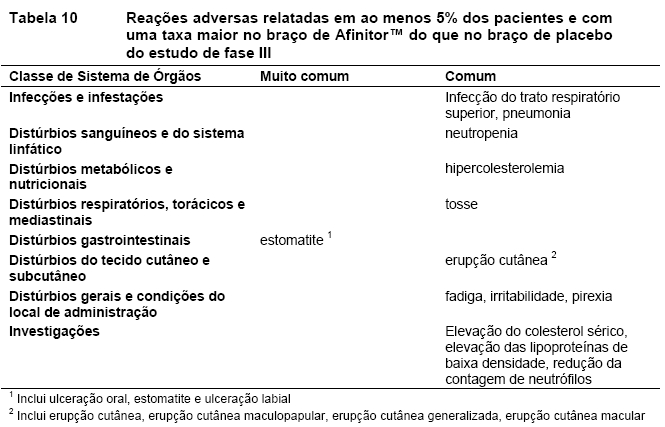
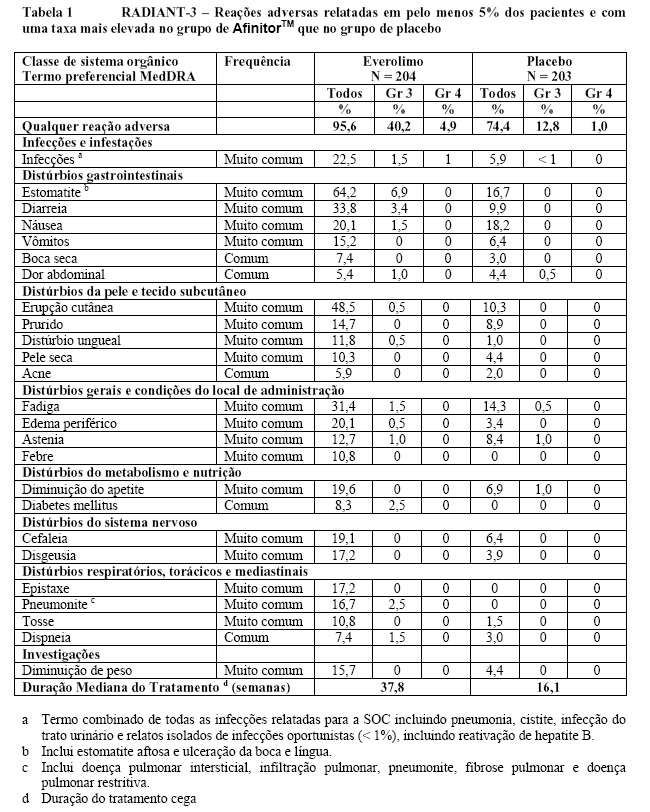
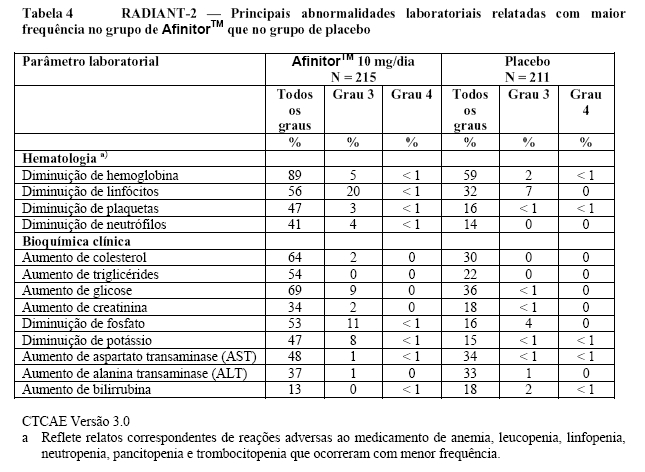
O RADIANT-3 (Estudo CRAD001C2324), um estudo de fase III, randomizado, duplo-cego e multicêntrico de AfinitorTM mais o "melhor tratamento de suporte" (BSC) versus placebo mais o BSC em pacientes com tumores neuroendócrinos pancreáticos (pNETs) avançados, demonstrou um benefício clínico estatisticamente significativo do AfinitorTM em relação ao placebo por um prolongamento de 2,4 vezes na sobrevida livre de progressão (SLP) mediana (11,04 meses versus 4,6 meses), resultando em uma redução de risco de 65% na SLP (RR de 0,35; IC de 95%: 0,27, 0,45; p < 0,0001)(veja a Tabela 1 e a Figura 1)1.
O RADIANT-3 incluiu pacientes com pNET avançado cuja doença havia progredido dentro dos 12 meses anteriores. Os pacientes foram estratificados por quimioterapia citotóxica anterior (sim/não) e por capacidade funcional de acordo com a OMS (0 vs. 1e 2). O tratamento com análogos da somatostatina foi permitido como parte do BSC.
O desfecho primário para o estudo foi a SLP avaliada pelos RECIST (Critérios de Avaliação de Resposta em Tumores Sólidos, versão1.0). Após a progressão radiológica documentada, os pacientes puderam ter a quebra do código cego pelo investigador: pacientes randomizados para o placebo puderam, então, receber o AfinitorTM aberto.
Os desfechos secundários incluem a segurança, a taxa de resposta objetiva (TRO) (resposta completa (RC) ou resposta parcial (RP)), a duração da resposta e a sobrevida geral (SG).
No total, 410 pacientes foram randomizados, a 1:1, para receber 10 mg/dia de AfinitorTM (n = 207) ou placebo (n = 203). Os dados demográficos foram bem equilibrados (idade mediana de 58 anos, 55% homens, 78,5% caucasianos).
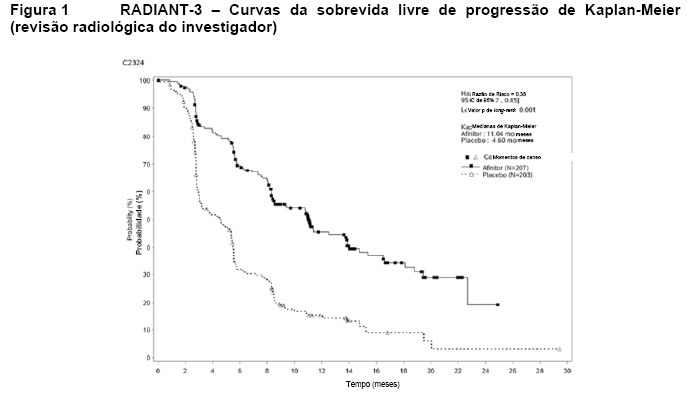
As taxas de SLP de dezoito meses foram de 34,2% para a terapia com AfinitorTM em comparação a 8,9% para o placebo.
A taxa de resposta objetiva de acordo com a avaliação do investigador foi de 4,8% para o braço de everolimo em comparação com 2,0% para o braço de placebo. A redução tumoral também foi evidente segundo o gráfico em cascata correspondente. Os resultados indicam que 64,4% dos pacientes no braço de everolimo apresentaram regressão tumoral em comparação com 20,6% no braço de placebo (Figura 2)12.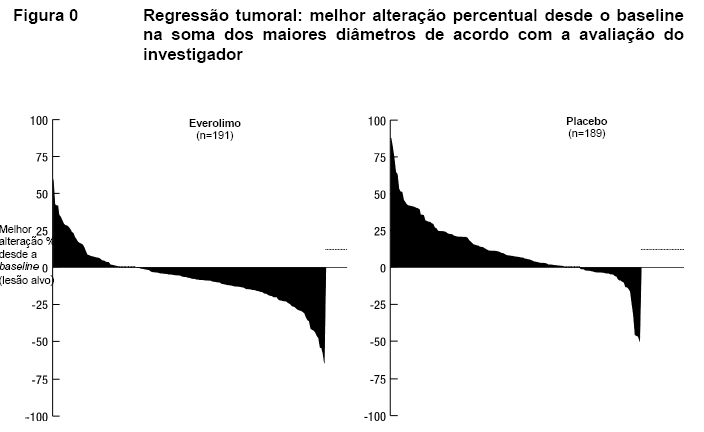

Os resultados da sobrevida geral ainda não estão inteiramente desenvolvidos e nenhuma diferença estatisticamente significativa na SG foi observada (RR = 0,99 (IC de 95% de 0,68 a 1,43) em uma análise atualizada). O cruzamento de ≥ 72% dos pacientes do placebo para o AfinitorTM aberto após a progressão da doença provavelmente confundiu a detecção de qualquer diferença relacionada ao tratamento na SG.
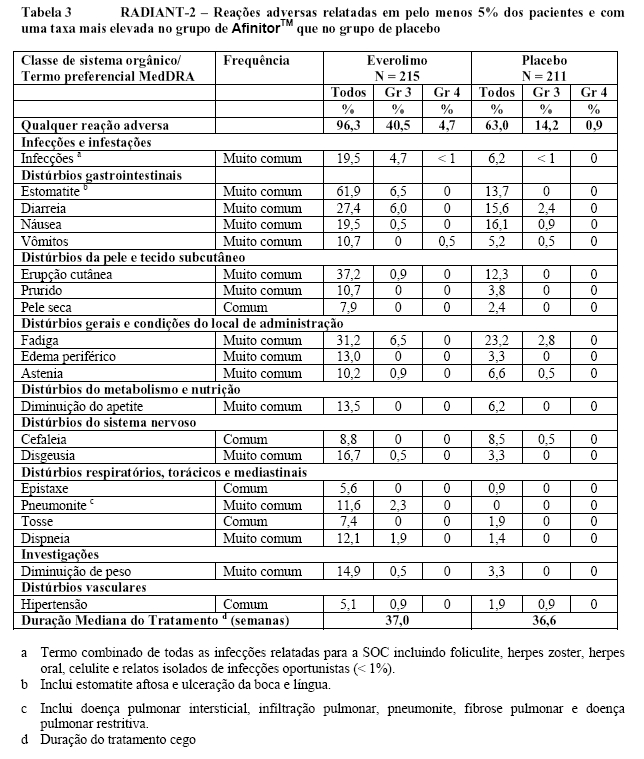
O RADIANT-2 (Estudo CRAD001C2325), um estudo de fase III, randomizado, duplo-cego e multicêntrico de AfinitorTM mais octreotida de depósito (Sandostatin LAR®) versus placebo mais octreotida de depósito em pacientes com tumores neuroendócrinos (tumor carcinoide) avançados primariamente de origem gastrintestinal ou pulmonar, mostrou evidências de benefício clínico do AfinitorTM em relação ao placebo por um prolongamento de 5,1 meses na SLP mediana (16,43 meses versus 11,33 meses; RR de 0,77; IC de 95%: 0,59 a 1,00; p = 0,026 unilateral), resultando em uma redução de risco de 23% na SLP primária (veja a Tabela 2 e a Figura 3). Embora a significância estatística não tenha sido alcançada para a análise primária (o limite para significância estatística foi de p = 0,0246), as análises que realizaram um ajuste em relação à censura informativa e aos desequilíbrios nos dois braços de tratamento mostraram um efeito do tratamento a favor do everolimo. O RADIANT-2 incluiu pacientes com tumores neuroendócrinos (tumor carcinoide) avançados, primariamente de origem gastrintestinal ou pulmonar, cuja doença havia progredido dentro dos 12 meses anteriores e que apresentavam histórico de sintomas secretores. 80,1% dos pacientes no grupo de AfinitorTM receberam uma terapia com análogo da somatostatina antes da entrada no estudo em comparação a 77,9% no grupo de placebo2.
O desfecho primário é a SLP avaliada pelo RECIST. Após a progressão radiológica documentada, os pacientes puderam ter a quebra do código cego pelo investigador: aqueles randomizados para o placebo puderam, então, receber o AfinitorTM aberto.
Os desfechos secundários incluem a segurança, resposta objetiva, a duração da resposta e a sobrevida geral.
No total, 429 pacientes foram randomizados, a 1:1, para receber 10 mg/dia de AfinitorTM (n = 216) ou placebo (n = 213), além de 30 mg de octreotida de depósito (Sandostatin LAR®, administrado intramuscularmente) a cada 28 dias. Desequilíbrios notáveis foram evidentes para diversos fatores prognósticos importantes de baseline, principalmente a favor do grupo de placebo.
As análises adicionais para a revisão radiológica independente, que realizaram um ajuste em relação à censura informativa e aos desequilíbrios nos dois braços de tratamento, mostraram um efeito do tratamento a favor do everolimo. Os resultados de uma análise multivariada ajustada adicional, que realizou uma correção em relação aos desequilíbrios entre os braços de tratamento, produziram uma RR de 0,73 (IC de 95% de 0,56 a 0,97). Um modelo de Cox com Probabilidade Inversa dos Pesos de Censura (IPCW) foi usado para abordar e realizar uma correção em relação à censura informativa e aos desequilíbrios nas características de baseline entre os dois braços do estudo. A RR estimada (IC de 95%) a partir da análise de IPCW foi de 0,60 (0,44 a 0,84), a favor do Afinitor™.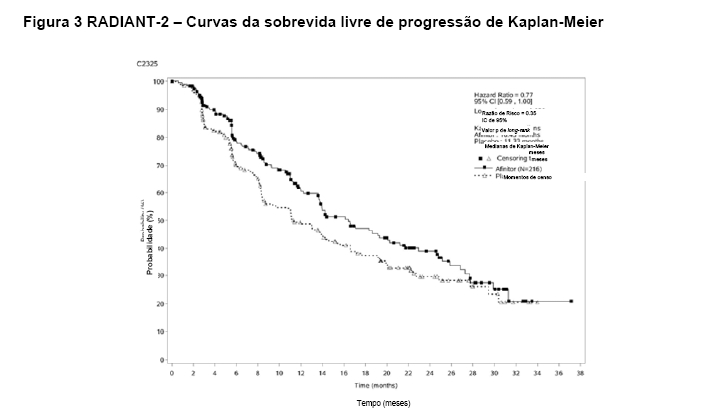
As taxas de SLP aos dezoito meses foram de 47,2% para a terapia com everolimo mais octreotida de depósito (Sandostatin LAR®) em comparação a 37,4% para placebo mais octreotida de depósito (Sandostatin LAR®).
A taxa de resposta objetiva de acordo com a avaliação central adjudicada foi de 2,3% para o braço de everolimo mais octreotida de depósito (Sandostatin LAR®) em comparação com 1,9% para o braço de placebo mais octreotida de depósito (Sandostatin LAR®). A redução tumoral também foi evidente segundo o gráfico em cascata correspondente. Os resultados indicam que 75,0% dos pacientes no braço de everolimo octreotida de depósito (Sandostatin LAR®) apresentaram regressão tumoral em comparação com 44,8% no braço de placebo mais octreotida de depósito (Sandostatin LAR®)12.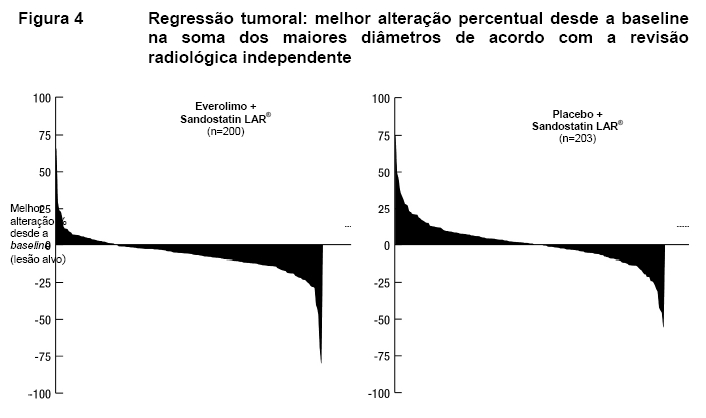

Os resultados da sobrevida global ainda não estão inteiramente desenvolvidos e nenhuma diferença estatisticamente significativa na SG foi observada (RR para a análise ajustada pré-especificada = 1,05 (IC de 95% de 0,79 a 1,39))13,14. O cruzamento de 58,2% (124/213) dos pacientes do placebo para o AfinitorTM aberto após a progressão da doença provavelmente confundiu a detecção de qualquer diferença relacionada ao tratamento na SG.
Carcinoma avançado de células renais
O RECORD-1 (Estudo CRAD001C2240), um estudo duplo-cego, randomizado, multicêntrico, internacional de fase III, foi conduzido comparando AfinitorTM 10 mg/dia e placebo, ambos em conjunto com o melhor tratamento de suporte em pacientes com carcinoma metastático de células renais cuja doença evoluiu apesar do tratamento prévio de terapia (sunitinibe, sorafenibe ou ambos sunitinibe e sorafenibe) com VEGFR-TKI (inibidor da quinase de tirosina do receptor do fator de crescimento endotelial vascular). A terapia prévia com bevacizumabe e alfainterferona também foi permitida. Os pacientes foram estratificados de acordo com critérios prognósticos do Memorial Sloan-Kettering Cancer Center (MSKCC) (grupos de risco favorável vs intermediário vs desfavorável) e terapia prévia anticâncer (1 vs 2 VEGFR-TKIs anteriores) 3,4.
A sobrevida livre de progressão, documentada usando RECIST (Critérios de Avaliação da Resposta em Tumores Sólidos) e avaliada por meio de uma análise central cega e independente, foi o objetivo primário. Os objetivos secundários incluíam segurança, taxa de resposta objetiva tumoral, sobrevida global, sintomas relacionados à doença e qualidade de vida. Após a progressão radiológica documentada, o investigador podia revelar o caráter cego aos pacientes: aqueles randomizados para placebo puderam receber 10 mg/dia de AfinitorTM em regime aberto. O Comitê Independente de Monitoramento de Dados recomendou o término deste estudo no momento da segunda análise interina assim que o objetivo primário foi alcançado3.
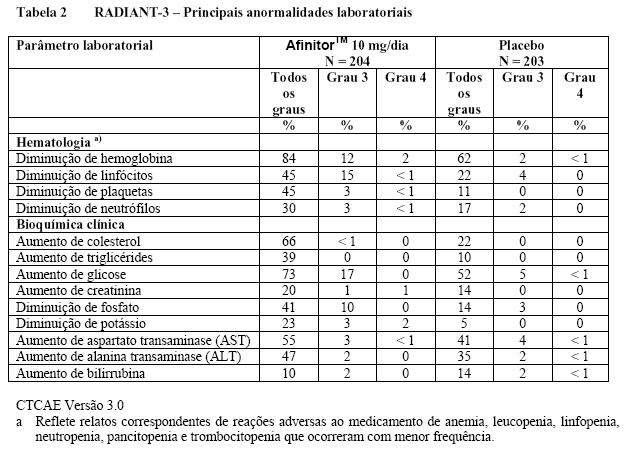
No total, 416 pacientes foram randomizados na proporção 2:1 para receber AfinitorTM (n = 277) ou placebo (n = 139). Os dados demográficos foram bem equilibrados (idade mediana agrupada de 61 anos [variação entre 27 a 85], 77% eram homens, 88% caucasianos e 74 % receberam pelo menos uma terapia prévia com VEGFR-TKI3,5,6. Os resultados da análise interina mostraram que o AfinitorTM foi superior ao placebo para o objetivo primário de sobrevida livre de progressão, com uma redução estatisticamente significativa de 67% no risco de progressão ou morte (vide Tabela 3 e Figura 5)3,5,6.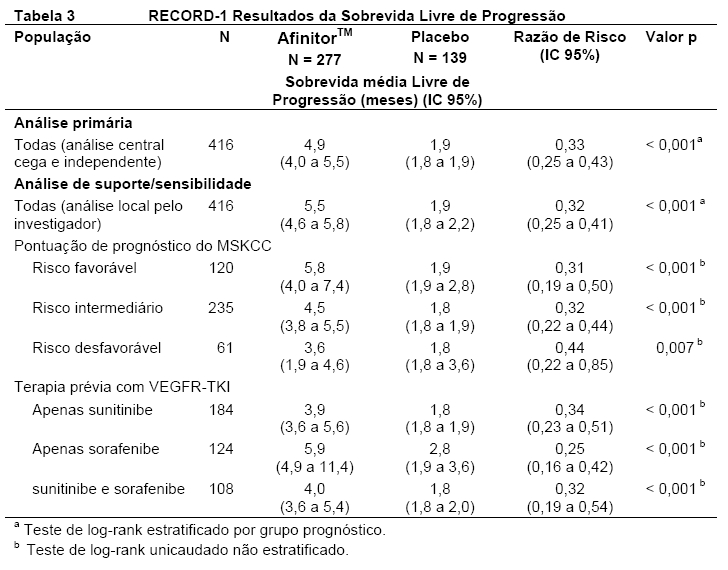
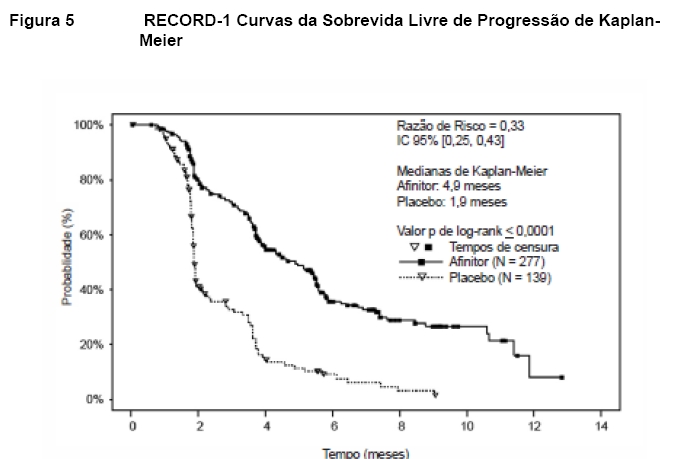
A taxa de sobrevida livre de progressão aos seis meses foi de 36 % para a terapia com AfinitorTM comparado com 9 % para placebo5,6. Respostas tumorais objetivas confirmadas foram observadas em 5 pacientes (2%) que receberam AfinitorTM enquanto nenhuma foi observada em pacientes que receberam placebo. Portanto, a vantagem da sobrevida livre de progressão reflete principalmente a população com estabilização da doença (correspondente a 67% do grupo de tratamento com AfinitorTM)5.
Não foi observada nenhuma diferença estatisticamente significativa relacionada ao tratamento na sobrevida global, apesar de ter havido uma tendência a favor do AfinitorTM (HR 0,82; 95% lC: 0,57 a 1,17; p = 0,137). O cruzamento com AfinitorTM em regime aberto após a progressão da doença para pacientes alocados para placebo confundiu a detecção de diferenças relacionadas ao tratamento na sobrevida global5,6. Uma melhora na qualidade de vida, medida através dos sintomas relacionados à doença, foi demonstrada nos pacientes que receberam AfinitorTM (HR 0,75; 95% IC: 0,53 a 1,06; p=0,053)5,6.
Esclerose Tuberosa com SEGA
Estudo de fase III em pacientes com TSC que possuem SEGA
O EXIST-1 (Estudo CRAD001M2301), um estudo de fase III, randomizado, duplo-cego, multicêntrico de Afinitor versus placebo, foi conduzido em pacientes com TSC e SEGA, independentemente da idade. Os pacientes foram randomizados a uma razão de 2:1 para receber Afinitor ou placebo correspondente. Era exigida a presença de ao menos uma lesão de SEGA ≥ 1,0 cm, no maior diâmetro, definido por ressonâcia magnética (com base na avaliação radiológica local) para a inclusão. Além disso, era necessária evidência radiológica em série de crescimento do SEGA, presença de uma nova lesão de SEGA ≥ 1 cm, no maior diâmetro, ou hidrocefalia nova ou agravada para a inclusão9,10,11,15.
O desfecho de eficácia primário foi uma taxa de resposta do SEGA baseada em uma análise radiológica central independente. A análise era estratificada pelo uso de medicamentos antiepiléticos indutores de enzimas (EIAEDs) na randomização (sim / não)9,10,11,15.
Os principais desfechos secundários, em ordem hierárquica de teste incluíram alteração absoluta na frequência do número total de eventos de crise epiléptica por meio de um EEG de 24 horas, desde a baseline até a Semana 24, tempo até a progressão do SEGA e taxa de resposta de lesões cutâneas9,10,11,15.
No total, 117 pacientes foram randomizados, sendo 78 para Afinitor e 39 para placebo. Os dois braços de tratamento estavam, bem balanceados em relação as características demográficas e da doença de baseline e o histórico de terapias anteriores anti- SEGA. A idade mediana foi de 9,5 anos de idade (variação: 0,8 a 26,6; 69,2% estavam entre 3 e < 18 anos de idade no momento da inclusão; 17,1% estava com < 3 anos de idade no momento da inclusão), 57,3% eram do sexo masculino e 93,2% eram caucasianos. Dos pacientes incluídos, 79,5% apresentavam SEGAs bilaterais, 42,7% apresentavam ≥ 2 lesões de SEGA, 25,6% apresentavam crescimento inferior, 9,4% apresentavam evidência de invasão profunda no parênquima, 6,8% apresentavam evidência radiológica de hidrocefalia e 6,8% haviam sido submetidos a cirurgia anterior relacionada à SEGA; 94,0% apresentavam lesões cutâneas na baseline e 37,6% apresentavam angiomiolipomas renais (ao menos um angiomiolipoma ≥ 1 cm no maior diâmetro). A duração mediana do tratamento do estudo cego foi de 9,6 meses (variação: 5,5 a 18,1) para os pacientes que receberam Afinitor e 8,3 meses (variação: 3,2 a 18,3) para aqueles que receberam placebo9,10,11,15.
Os resultados demonstram que Afinitor foi superior ao placebo com relação ao desfecho primário de melhor resposta geral do SEGA (p < 0,0001). As taxas de resposta foram 34,6% (IC de 95%: 24,2, 46,2) para o braço de Afinitor comparado a 0% (IC de 95%: 0,0, 9,0) para o braço de placebo (Tabela 4). Além disso, todos os 8 pacientes do braço de Afinitor que apresentaram evidência radiológica de hidrocefalia na baseline apresentaram redução no volume ventricular e nenhuma paciente necessitou de intervenção cirúrgica durante o decorrer deste estudo9,10,11,15.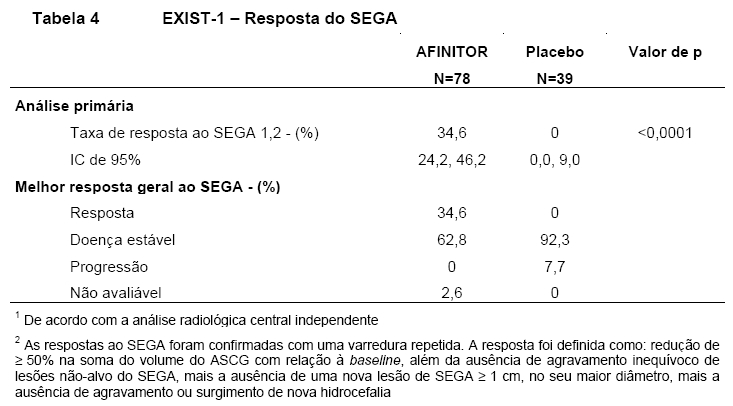
Foram observados efeitos consistentes com o tratamento em todos os subgrupos avaliados (ou seja, uso de EIAED versus sem o uso de EIAED, sexo e idade) (Tabela 5, Figura 6) 9,10,11,15.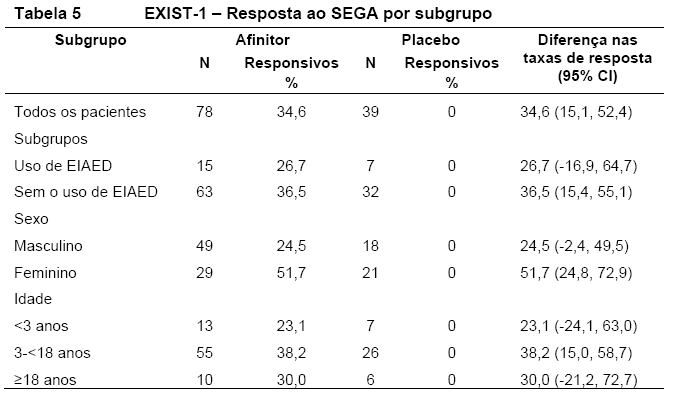
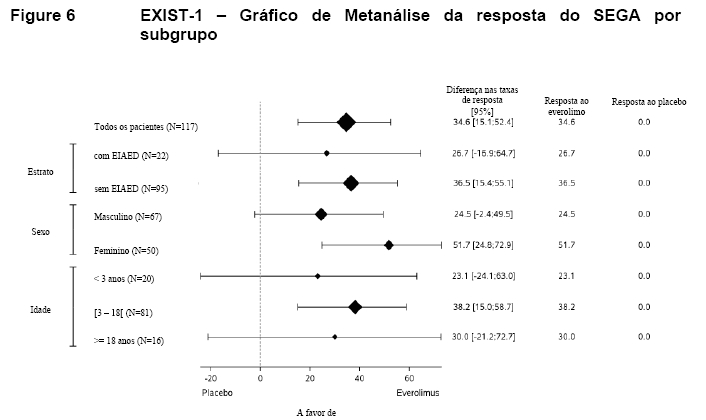
Nas primeiras 12 semanas de tratamento com Afinitor, a redução do SEGA foi evidente: 73,0% dos pacientes apresentaram reduções de ≥ 30% e 29,7% apresentaram reduções de ≥ 50% na ocasião da primeira avaliação radiológica (Semana 12). Nos períodos subsequentes, reduções consistentes foram evidentes; na Semana 24, 78,4% dos pacientes apresentaram reduções ≥ 30% e 41,9% apresentaram reduções ≥ 50%9,10,11.
A análise do primeiro desfecho secundário principal, alteração da frequência de crises epilépticas, foi inconclusiva.
O tempo mediano até a progressão do SEGA com base na análise radiológica central não foi alcançado em nenhum braço de tratamento. Foram somente observadas progressões no braço de placebo (15,4%; p=0,0002) (Figura 7). As taxas estimadas livres de progressão no sexto mês foram de 100% para o braço de Afinitor e 85,7% para o braço de placebo9,10,11,15.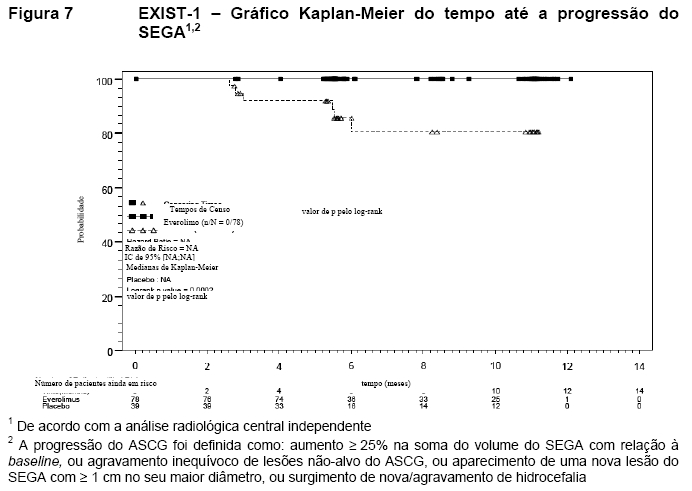
Afinitor demonstrou melhora clinicamente significativa na resposta de lesões cutâneas (p=0,0004), com taxas de resposta de 41,7% (IC de 95%: 30,2, 53,9) para o braço de Afinitor e 10,5% (IC de 95%: 2,9, 24,8) para o braço de placebo (Tabela 6)9,10,11,15.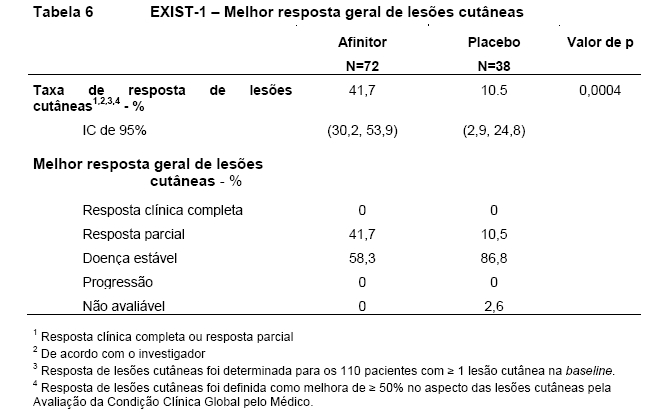
Estudos fase II em pacientes com TSC e SEGA
Um estudo prospectivo, aberto, de fase II foi conduzido para avaliar a segurança e a eficácia de AfinitorTM em pacientes com SEGA. Evidências radiológicas seriadas do crescimento do SEGA foram exigidas para inclusão7, 8.
A alteração no volume do SEGA durante a fase de tratamento central de 6 meses, avaliada por uma revisão radiológica central independente, constituiu o desfecho primário de eficácia. Após a fase de tratamento central, os pacientes poderiam entrar na fase de tratamento de extensão, onde o volume do SEGA foi avaliado a cada 6 meses7,8.
No total, 28 pacientes receberam tratamento com AfinitorTM; a idade mediana correspondeu a 11 anos (variação de 3 a 34), 61% do sexo masculino, 86% caucasianos. Treze pacientes (46%) apresentaram um SEGA secundário menor, incluindo 12 pacientes com SEGA no ventrículo contralateral8.
AfinitorTM foi associado a uma redução clinicamente relevante e estatisticamente significativa no volume do SEGA primário após 6 meses em relação à avaliação basal (p < 0,001). A redução do tumor foi mais rápida durante os 3 meses de tratamento iniciais, com evidência de uma resposta mantida em pontos de tempo subsequentes (ver Tabela 7). Nenhum paciente desenvolveu novas lesões, agravamento de hidrocefalia, aumento da pressão intracraniana e nenhum necessitou de ressecção cirúrgica ou outra terapia para SEGA7, 8.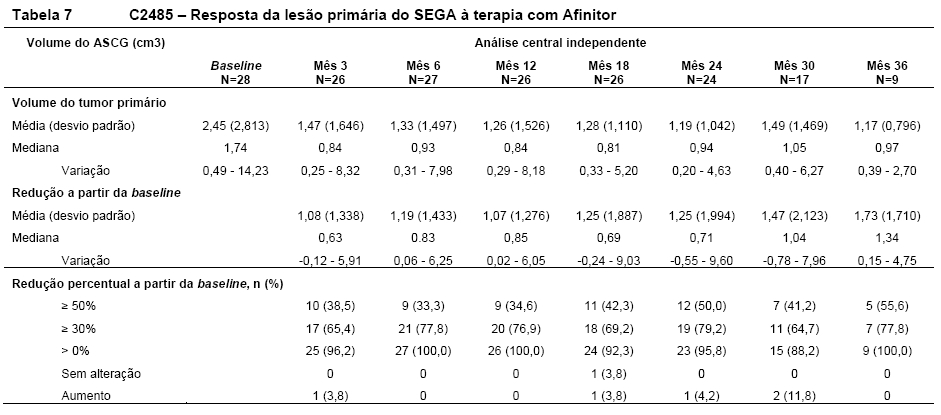
A análise primária foi confirmada pela:
• Alteração no volume do SEGA primário conforme a avaliação local do investigador (p < 0,001), com 75% e 39% dos pacientes apresentando reduções ≥ 30% e ≥ 50%, respectivamente.
• Alteração no volume total do SEGA conforme a revisão central independente (p < 0,001) ou a avaliação local do investigador (p < 0,001)
Um paciente satisfez os critérios pré-especificados de sucesso terapêutico (redução > 75% no volume do SEGA) e foi descontinuado temporariamente da terapia de estudo; contudo, o novo crescimento do SEGA foi evidente dentro de 3 meses e o tratamento foi reiniciado.
Acompanhamento em longo prazo de duração mediana de 34,2 meses (faixa: 4,7 a 47,1) demonstrou eficácia sustentada9,10,11,15.
Referências
1. [Study C2324] A randomized double-blind phase III study of RAD001 10 mg/d plus best supportive care versus placebo plus best supportive care in the treatment of patients with advanced pancreatic neuroendocrine tumor (NET). Novartis Pharmaceuticals Corporation. East Hanover, USA. 29-Aug-2010 [74] (dados em arquivo)
2. [Study C2325] A randomized, double-blind, placebo-controlled, multicenter phase III study in patients with advanced carcinoid tumor receiving Sandostatin LAR® Depot and RAD001 10 mg/d or Sandostatin LAR® Depot and placebo. Novartis Pharmaceuticals Corporation. East Hanover, USA. 01-Oct-2010. [75] (dados em arquivo)
3. [Study C2240] A randomized, double-blind, placebo-controlled, multicenter phase III study to compare the safety and efficacy of RAD001 plus Best Supportive Care (BSC) versus BSC plus Placebo in patients with metastatic carcinoma of the kidney which has progressed on VEGF receptor tyrosine kinase inhibitor therapy. Full Clinical Study Report RAD001 C2240. Novartis Pharmaceuticals Corporation. East Hanover, USA. 29 May 08 [2] (dados em arquivo)
4. [Summary of Clinical Efficacy] RAD001 (everolimus) - 2.7.3. Summary of Clinical Efficacy in advanced renal cell carcinoma. Novartis Pharmaceuticals Corporation. East Hanover, USA. 28 May 08. [44] (dados em arquivo).
5. [Study C2240] A randomized, double-blind, placebo-controlled, multicenter phase III study to compare the safety and efficacy of RAD001 plus Best Supportive Care (BSC) versus BSC plus Placebo in patients with metastatic carcinoma of the kidney which has progressed on VEGF receptor tyrosine kinase inhibitor therapy. Full Clinical Study Report-Addendum Report RAD001 C2240. Novartis Pharmaceuticals Corporation. East Hanover, USA. [51] (dados em arquivo)
6. Expert Statement - Update to Warning and Precautions and Adverse Drug Reactions. 02 Apr 09. [54] (dados em arquivo)
7. [Clinical Overview] in Subependymal giant cell astrocytoma associated with tuberous sclerosis. Novartis Pharmaceuticals Corporation. East Hanover, USA. 31-Mar-10.
8. [Study C2485] Everolimus (RAD001) Therapy of Giant Cell Astrocytoma in Patients with Tuberous Sclerosis Complex. Clinical Study Report RAD001 C2485. Novartis Pharmaceuticals Corporation. East Hanover, USA. 31-Mar-10.
9. [Clinical Overview] Tuberous sclerosis complex with subependymal giant cell astrocytoma. Novartis Pharmaceuticals Corporation. East Hanover, USA. 02-Feb-2012
10. [Study M2301] A randomized, double-blind, placebo-controlled study of RAD001 in the treatment of patients with subependymal giant cell astrocytomas (SEGA) associated with tuberous sclerosis complex (TSC). Novartis Pharmaceuticals Corporation. East Hanover, USA. 14-Sep-2011.
11. [Summary of Clinical Efficacy] Tuberous sclerosis complex with subependymal giant cell astrocytoma. Novartis Pharmaceuticals Corporation. East Hanover, USA. 12-Jan-2012.
12. Study C2324] A randomized double-blind phase III study of RAD001 10 mg/d plus best supportive care versus placebo plus best supportive care in the treatment of patients with
advanced pancreatic neuroendocrine tumor (NET). Novartis Pharmaceuticals Corporation. East Hanover, USA. 29-Aug-2010
13. [Clinical Overview]. Advanced Neuroendocrine Tumors. Novartis Pharmaceuticals Corporation. East Hanover, USA. 22-Oct-2010
14. [Errata to Clinical Overview]. Errata to Clinical Overview in Advanced Neuroendocrine Tumors. Novartis Pharmaceuticals Corporation. East Hanover, USA. 10-Dec-2010.
15. [Clinical Overview] Tuberous sclerosis complex with subependymal giant cell astrocytoma. Novartis Pharmaceuticals Corporation. East Hanover, USA. 19-Mar-2012
Caract. farmacológicas.
Grupo farmacoterapêutico: inibidor de proteina-quinase, código ATC: L01XE10
Mecanismo de ação
O everolimo é um inibidor de transdução de sinal com alvo no mTOR (alvo da rapamicina de mamíferos), ou mais especificamente, mTORC1 ('alvo da rapacimina' complexo 1 de mamíferos). O mTOR é a principal serina-treonina quinase que desempenha função central na regulação do crescimento, proliferação e sobrevida celular. A regulação da sinalização do mTORC1 é complexa, sendo modulada por mitógenos, fatores de crescimento, disponibilidade de energia e nutriente. O mTORC1 é um regulador essencial da síntese protéica downstream global na via PI3K/AKT, que está desregulada na maioria dos cânceres humanos.
Dois reguladores primários da sinalização de mTORC1 são os oncogenes supressores do complexo esclerose-tuberina 1 & 2 (TSC1, TSC2). A perda ou a inativação de TSC1 ou TSC2 provoca uma elevação dos níveis de rheb-GTP, uma GTPase da família ras, que interage com o complexo mTORC1 causando sua ativação. A ativação de mTORC1 provoca uma cascata subsequente de sinalização de quinases, incluindo a ativação de S6K1. No complexo da esclerose tuberosa, um distúrbio genético, mutações de inativação no gene TSC1 ou TSC2 promovem a formação de hamartomas em todo o organismo.
Propriedades farmacodinâmicas
O everolimo é um inibidor seletivo de mTOR (alvo da rapamicina de mamíferos), com alvo específico no complexo de transdução do sinal de mTOR-raptor (mTORC1). O mTOR é a principal serina-treonina quinase na cascata de sinalização PI3K/AKT, uma via conhecida por ser desregulada na maioria dos cânceres humanos. O everolimo exerce sua atividade por meio de uma interação de alta afinidade com a proteína do receptor intracelular, FKBP12. O complexo FKBP12/everolimo liga-se ao mTORC1, inibindo sua capacidade de sinalização. A sinalização do mTORC1 é afetada pela modulação da fosforilação dos efetores downstream, sendo que o melhor caracterizado é a proteína quinase ribossômica dos reguladores de tradução S6 (S6K1) e a proteína de ligação 4E do fator de alongamento eucariótico (4E-BP). O rompimento da função S6K1 e 4E-BP1, como consequência da inibição do mTORC1, interfere na tradução dos mRNAs que codificam as proteínas pivotais envolvidas na regulação do ciclo celular, glicólise e adaptação a baixas condições de oxigênio (hipóxia). Isso inibe o crescimento do tumor e a expressão de fatores de indução de hipóxia (por exemplo, fatores de transcrição de HIF-1); o último resultando na redução de expressão de fatores envolvidos na potencialização dos processos angiogênicos do tumor (por exemplo, o fator de crescimento endotelial vascular, VEGF). O everolimo é um potente inibidor do crescimento e proliferação de células tumorais, células endoteliais, fibroblastos e células do músculo liso associadas ao vaso sanguíneo. De forma consistente com a função reguladora central do mTORC1, o everolimo demonstrou reduzir a proliferação da célula tumoral, glicólise e angiogênese em tumores sólidos in vivo, e assim propicia dois mecanismos independentes para inibição do crescimento tumoral: a atividade antitumoral direta e a inibição do estroma tumoral.
Em um modelo neuronal murino de TSC, com a ablação de TSC1 na maioria dos neurônios durante o desenvolvimento cortical, everolimo elevou a sobrevida mediana de 33 dias para mais de 100 dias, além de uma melhora acentuada do comportamento, do fenótipo e do ganho de peso. Houve boa penetração cerebral, com acúmulo ao longo do tempo após tratamento repetitivo e redução efetiva dos níveis de fosfo-S6, um marcador subsequente de mTORC1. As anormalidades de neurofilamentos, mielinização e aumento celular melhoraram com o tratamento, embora características neuronais displásicas tenham persistido, e houve alterações apenas modestas na densidade e comprimento das espinhas dendríticas. Camundongos tratados com everolimo apenas por 23 dias (dias pós-natais 7-30) exibiram uma melhora persistente do fenótipo, com uma sobrevida mediana de 78 dias. Em resumo, everolimo é muito ativo neste modelo neuronal de TSC, com o benefício aparentemente atribuível aos efeitos sobre a sinalização de mTORC1 e Akt e, consequentemente, o tamanho celular e a mielinização.
Propriedades farmacocinéticas
Absorção
Em pacientes com tumores sólidos avançados, as concentrações de pico de everolimo são alcançadas de 1 a 2 horas após a administração de uma dose oral de 5 a 70 mg de everolimo em jejum ou com uma refeição leve sem gordura. A Cmáx é proporcional à dose entre 5 e 10 mg em regimes diários e semanais. Em doses de 20 mg/semana e superiores, o aumento na Cmáx é menor do que aquela Cmáx proporcional à dose, porém a AUC demonstra uma proporcionalidade à dose com relação à variação da dose de 5 a 70 mg.
Efeito do alimento: Em indivíduos sadios, uma refeição com alto teor de gordura reduziu em 22% a exposição sistêmica de AfinitorTM 10 mg (medida através da AUC) e em 54%, o pico de concentração plasmática. Refeições com baixo teor de gordura reduziram a AUC e Cmáx em 32 % e em 42%, respectivamente. Entretanto, o alimento não apresenta efeito aparente no perfil concentração versus tempo após a absorção.
Distribuição
A razão sangue-plasma de everolimo, que é concentração-dependente da variação de 5 a 5.000 ng/mL, é de 17% a 73%. A quantidade de everolimo restrita ao plasma é de aproximadamente 20% nas concentrações séricas observadas em pacientes com câncer que receberam AfinitorTM 10 mg/dia. A ligação protéica no plasma é de aproximadamente 74% tanto em indivíduos sadios quanto em pacientes com insuficiência hepática moderada.
Após a administração intravenosa em um modelo de rato, o everolimo demonstrou atravessar a barreira hemato-encefálica de forma dose-dependente não linear, sugerindo a saturação da bomba de efluxo na barreira hemato-encefálica. A penetração de everolimo no cérebro também foi demonstrada em ratos que receberam doses orais de everolimo.
Metabolismo
O everolimo é um substrato da CYP3A4 e PgP. Após a administração oral, ele é o principal componente na circulação do sangue humano. Seis metabólitos principais de everolimo foram detectados no sangue humano, incluindo três metabólitos monohidroxilados, dois produtos hidrolíticos com anel aberto e um conjugado fosfatidilcolina do everolimo. Esses metabólitos também foram identificados em espécies animais usadas em estudos de toxicidade e demonstraram atividade aproximadamente 100 vezes menor do que o everolimo. Por essa razão, considera-se que a substância inicial contribua para a maioria da atividade farmacológica geral do everolimo.
Excreção
Não foi realizado nenhum estudo específico de excreção em pacientes com câncer; no entanto, estão disponíveis dados do cenário de transplante. Após a administração de uma única dose de everolimo marcado com rádio juntamente com ciclosporina, 80% da radioatividade foi recuperada das fezes, enquanto 5% foi excretado na urina. A substância inicial não foi detectada na urina ou nas fezes.
Farmacocinética no estado de equilíbrio
Após a administração diária ou semanal de everolimo em pacientes com tumores sólidos avançados, a AUC0-T no estado de equilíbrio foi proporcional à dose na variação de 5 a 10 mg no regime de dosagem diária e de 5 a 70 mg no regime de dosagem semanal. O estado de equilíbrio foi alcançado em duas semanas com o regime de dosagem diária. A Cmáx é proporcional à dose entre 5 e 10 mg nos regimes diário e semanal. Em doses de 20 mg/semana e superiores, o aumento na Cmáx é menor do que aquela Cmáx proporcional à dose. O tmáx ocorre de 1 a 2 horas após a dose. Houve uma correlação significativa entre a AUC0-T e a concentração no vale na pré-dose no estado de equilíbrio no regime diário. A meia-vida de eliminação média do everolimo é de aproximadamente 30 horas.
Dados de segurança pré-clínica
O perfil de segurança pré-clínica de everolimo foi avaliado em camundongos, ratos, mini-porcos, macacos e coelhos. Os principais órgãos-alvo foram os sistemas reprodutivos de machos e fêmeas (degeneração tubular testicular, redução da quantidade de esperma nos epidídimos e atrofia uterina) em várias espécies; pulmões (aumento dos macrófagos alveolares) em ratos e camundongos; e olhos (opacidades lenticulares anteriores do fio de sutura) apenas nos ratos. Alterações menores nos rins foram observadas nos ratos (exacerbação da lipofuscina relacionada à idade no epitélio tubular, aumentos na hidronefrose) e camundongo (exacerbação das lesões subjacentes). Não houve indicação de toxicidade renal em macacos ou mini-porcos. O everolimo pareceu exacerbar espontaneamente doenças subjacentes (miocardite crônica em ratos, infecção pelo vírus de coxsackie do plasma e coração em macacos, infestação por coccidian do trato gastrintestinal em mini-porcos, lesões cutâneas em camundongos e macacos). Esses achados foram geralmente observados nos níveis de exposição sistêmica na variação da exposição terapêutica ou acima, com exceção dos achados em ratos, que ocorreram abaixo da exposição terapêutica devido à elevada distribuição no tecido. Em um estudo de fertilidade em ratos machos, a morfologia testicular foi afetada com 0,5 mg/kg e acima, e a mobilidade do esperma, contagem de espermas e níveis de testosterona no plasma diminuíram com 5 mg/kg, o que está dentro da variação de exposição terapêutica e que causou uma redução na fertilidade dos machos. Houve uma evidência de reversibilidade. A fertilidade das fêmeas não foi afetada, mas o everolimo atravessou a placenta e foi tóxico ao feto. Em ratos, o everolimo causou toxicidade embrionária/fetal na exposição sistêmica abaixo do nível terapêutico. Isso foi manifestado como mortalidade e redução do peso do feto. A incidência de variações esqueléticas e más-formações (por exemplo, fenda do esterno) aumentou em 0,3 e 0,9 mg/kg. Em coelhos, a embriotoxicidade foi evidente em um aumento de reabsorções tardias. Em estudos de toxicidade juvenil em ratos com doses baixas de 0,15 mg/kg/dia, a toxicidade sistêmica incluiu diminuição do ganho de peso corporal e do consumo de alimentos e retardo da obtenção de alguns marcos do desenvolvimento em todas as doses, com recuperação total ou parcial após o fim da administração. Com a possível exceção de achados no cristalino específicos para ratos (onde os animais jovens pareceram ser mais susceptíveis), parece não haver uma diferença significativa na sensibilidade de animais jovens aos efeitos adversos de everolimo em comparação aos animais adultos com doses de 0,5 a 5 mg/kg por dia. Nenhuma toxicidade relevante foi evidente em macacos jovens com doses de até 0,5 mg/kg/dia por 4 semanas. Estudos de genotoxicidade que incluem objetivos relevantes de genotoxicidade não mostraram nenhuma evidência de atividade clastogênica ou mutagênica. A administração de everolimo por até 2 anos não indicou nenhum potencial oncogênico em camundongos e ratos até as doses mais elevadas, correspondentes, respectivamente, a 3,9 e 0,2 vezes a exposição clínica estimada.
Contraindicações.
Hipersensibilidade ao princípio ativo, a outros derivados da rapamicina ou a qualquer um dos excipientes (vide "Advertências" e "Precauções").
Advertências e precauções.
Advertências
Uso em idosos, crianças e outros grupos de risco
Pacientes pediátricos:
- AfinitorTM não está recomendado para a população pediátrica com carcinoma avançado de células renais. (vide "Posologia").
• Em pacientes com TSC e SEGA, a Cmin de everolimo foi aproximadamente proporcional a dose com a faixa de dosagem de 1,35 mg/m2 a 14,4 mg/m2.
• Em pacientes com TSC e SEGA, a mediana geométrica dos valores de Cmín, normalizados para uma dose em mg/m2, em pacientes com < 10 anos de idade e entre 10 e 18 anos, foi estatisticamente inferior à observada em adultos ( > 18 anos de idade), o que sugere que o clearance de everolimo foi maior em pacientes mais jovens.
Pacientes Idosos
Na avaliação farmacocinética da população em pacientes com câncer, nenhuma influência significativa da idade (27 a 85 anos) sobre o clearance (depuração) oral (CL/F: variação de 4,8 a 54,5 Litros/hora) de everolimo foi detectada.
Etnia
O clearance (depuração) oral (CL/F) é semelhante em pacientes japoneses e caucasianos com câncer com funções hepáticas semelhantes. Com base na análise farmacocinética da população, o clearance (depuração) oral (CL/F) é em média 20% mais elevado em pacientes negros transplantados.
Pacientes com insuficiência hepática
A segurança, tolerabilidade e farmacocinética do Afinitor foram avaliadas em um estudo de dose oral única de everolimo em 34 indivíduos com função hepática comprometida em relação a indivíduos com função hepática normal. Em comparação com indivíduos normais, houve um aumento de 1,6 vezes, 3,3 vezes e 3,6 vezes na exposição (ou seja, AUC(0-inf)) para indivíduos com grau leve (Child-Pugh A), moderada (Child-Pugh B) e insuficiência hepática grave (Child-Pugh C), respectivamente. Simulações de farmacocinética de doses múltiplas de apoio as recomendações de dose em indivíduos com comprometimento hepático com base em seu status Child Pugh. O ajuste de dose é recomendado para pacientes com insuficiência hepática (vide "Advertências e Precauções" e "Posologia e Administração").
Pacientes com insuficiência renal
Em uma análise farmacocinética da população de 170 pacientes com câncer avançado, nenhuma influência significativa do clearance (depuração) da creatinina (25 a 178 mL/min) foi detectada sobre o CL/F do everolimo. A insuficiência renal pós-transplante (variação do clearance (depuração) da creatinina entre 11 e 107 mL/min) não afetou a farmacocinética do everolimo em pacientes transplantados.
Relações exposição-resposta
Houve uma correlação moderada entre a diminuição na fosforilação de 4E-BP1 (P4EBP1) no tecido tumoral e a Cmín média do everolimo no estado de equilíbrio no sangue após a administração diária de 5 ou 10 mg de everolimo. Outros dados sugerem que a inibição da fosforilação da quinase S6 é muito sensível à inibição do mTOR pelo everolimo. A inibição da fosforilação do elF-4G foi completa em todos os valores de Cmín após a dose diária de 10 mg. Em pacientes com TSC e SEGA, uma análise baseada no modelo indicou que uma elevação duas vezes maior da Cmín levou a uma redução tumoral de 13% (IC de 95%: -18,2%, -7,5%) a partir da baseline, o que foi estatisticamente significante a um nível de 5%. Uma tendência sugestiva de uma sobrevida livre de progressão mais longa, com uma Cmin normalizada pelo tempo mais alto do everolimo (definida como [área sobre a curva Cmin-tempo a partir do início do estudo até o tempo do evento]/[tempo do início do estudo até o evento]), foi evidente em pacientes com tumores neuroendócrinos pancreáticos avançados (pNETs, razão de risco de 0,73; IC de 95%: 0,50 a 1,08) e em pacientes com tumor carcinoide avançado (razão de risco de 0,66; IC de 95%: 0,40 a 1,08). A Cmin do everolimo impactou a probabilidade de redução do tamanho do tumor (p < 0,001), com as razões de probabilidades de 1,62 e 1,46, respectivamente, para uma alteração na exposição de 5 ng/mL para 10 ng/mL em pacientes com pNET avançado e em pacientes com tumor carcinoide avançado.
Gravidez
AfinitorTM enquadra-se na categoria C de risco na gravidez.
Não existem dados adequados sobre o uso de AfinitorTM em mulheres grávidas. Estudos em animais mostraram efeitos da toxicidade reprodutiva incluindo toxicidade embrionária e toxicidade fetal (vide "Dados de segurança pré-clínica"). O risco potencial para humanos é desconhecido. AfinitorTM não deve ser administrado em mulheres grávidas, a menos que o benefício potencial supere o risco potencial ao feto.
Mulheres com potencial para engravidar
Mulheres com potencial para engravidar devem ser aconselhadas a usar um método contraceptivo altamente eficaz enquanto estiverem tomando AfinitorTM , e por até 8 semanas após o fim do tratamento.
Amamentação
Não se sabe se o everolimo é excretado no leite materno. Entretanto, em estudos com animais, everolimo e/ou seus metabólitos passaram rapidamente para o leite de ratas lactantes. Portanto, mulheres que tomam AfinitorTM não devem amamentar.
Fertilidade
O potencial de everolimo em causar infertilidade em pacientes homens e mulheres não é conhecido. Entretanto, foi observada amenorreia (inclusive amenorreia secundaria). Com base em achados não-clínicos, a fertilidade masculina pode ser comprometid