IQIRVO
IPSEN
elafibranor
Tratamento da colangite biliar primária.
Apresentações.
Comprimidos revestidos de 80 mg.
Frasco PEAD (frasco plástico) com tampa de rosca resistente a crianças com unidade dessecante integrada contendo 30 comprimidos revestidos.
USO ORAL
USO ADULTO
Composição.
Cada comprimido revestido contém: elafibranor 80 mg. Excipientes: celulose microcristalina, povidona, croscarmelose sódica, dióxido de silício coloidal, estearato de magnésio, Opadry® laranja (álcool polivinílico-parcialmente hidrolisado, dióxido de titânio, macrogol, talco, óxido de ferro amarelo, óxido de ferro vermelho).
Informações técnicas.
1. INDICAÇÕES
IQIRVO® é indicado para o tratamento da colangite biliar primária (CBP) em combinação com ácido ursodesoxicólico (AUDC) em adultos com uma resposta inadequada ao AUDC, ou como monoterapia em adultos incapazes de tolerar o AUDC.
2. RESULTADOS DE EFICÁCIA
A eficácia de elafibranor foi avaliada no Estudo GFT505B-319-1 (ELATIVE), um estudo fase 3, randomizado, duplo-cego, placebo-controlado seguido por uma extensão de longo prazo aberta em 161 adultos com CBP com uma resposta inadequada ou intolerância ao AUDC. Os participantes foram randomizados numa razão 2:1 para receber elafibranor 80 mg ou placebo uma vez ao dia por pelo menos 52 semanas. Quando aplicável, os participantes continuaram na sua dose de AUDC pré-estudo ao longo do estudo. Os participantes foram incluídos no estudo se sua fosfatase alcalina (FA) fosse ≥ 1,67 x o limite superior da normalidade (LSN) e a bilirrubina total (BT) fosse ≤ 2 x LSN. Os participantes foram excluídos no caso de cirrose descompensada ou outras causas de doença hepática.
No geral, a idade média foi 57,1 anos e o peso médio foi 70,8 kg. A população do estudo foi predominantemente feminina (96%) e branca (91%). A concentração média basal de FA foi 321,9 U/L e 39% dos participantes tinham uma concentração basal de FA > 3 x LSN.
A concentração média basal de BT foi 9,6 mmol/L e 96% dos participantes tinham uma concentração basal de BT menor ou igual ao LSN. A média basal da medida de rigidez hepática (LSM) por elastografia transitória foi 10,1 kPa. A pontuação média basal na PBC Worst Itch Numeric Rating Scale (NRS) foi 3,3 e 41% tinham prurido moderado a grave no início do estudo (pontuação na escala PBC Worst Itch NRS ≥4); para aqueles com prurido moderado a grave, a pontuação média basal na escala PBC Worst Itch NRS foi 6,2 para os participantes no grupo elafibranor 80 mg e 6,3 para participantes no grupo placebo. A maioria (95%) dos participantes recebeu tratamento em combinação com AUDC ou como monoterapia em 5% dos participantes que eram incapazes de tolerar o AUDC.
O desfecho primário foi resposta a colestase na semana 52, definido como desfecho composto: FA < 1,67 x LSN e BT ≤ LSN e diminuição da FA ≥ 15%. Os desfechos secundários principais foram normalização da FA na semana 52 e a alteração do prurido em relação ao inicial até a semana 52 e até a semana 24, baseado na pontuação na escala PBC Worst Itch NRS nos participantes com prurido moderado a grave no início do estudo.
Tabela 1 mostra o desfecho primário composto da resposta à colestase e o desfecho secundário principal de normalização da FA.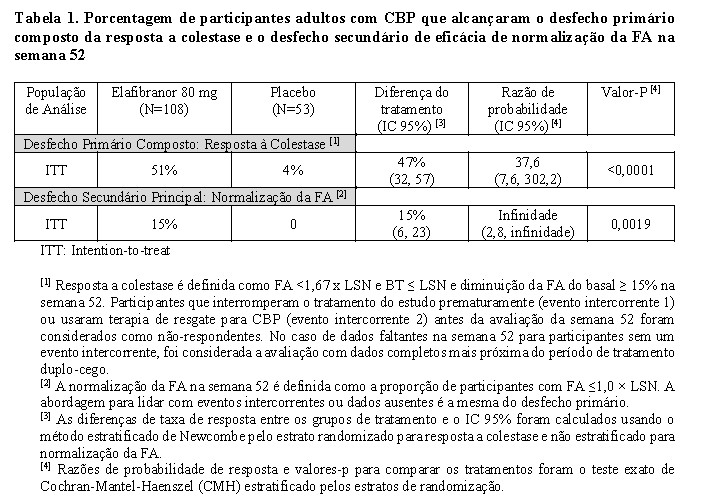
Uma diminuição significante da fosfatase alcalina (FA) em relação ao início foi vista a partir da semana 4 e foi mantida ao longo das 52 semanas de tratamento no grupo elafibranor comparada ao placebo. (Figura 1).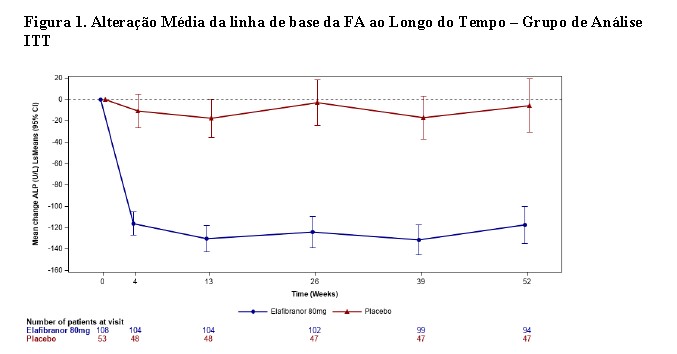
O desfecho primário de resposta bioquímica em participantes com FA basal ≤ 3 x o LSN foi alcançado em 71% dos participantes tratados com elafibranor, versus 6% dos participantes com placebo (AUDC), em comparação com aqueles com FA > 3x o LSN, nos quais a resposta bioquímica foi alcançada em 21% dos participantes tratados com elafibranor versus 0% com placebo.
Entre os 54 participantes com doença avançada, 16/35 (46%) dos participantes tratados com elafibranor versus 0/19 (0%) com placebo alcançaram o desfecho primário de resposta bioquímica. Devido ao número limitado de participantes com doença avançada, esses resultados devem ser interpretados com cautela.
Resultados relatados pelo paciente
Em participantes com prurido moderado a grave no início do estudo, a alteração média em relação ao início na escala PBC Worst Itch NRS até a semana 52 e semana 24 diminuiu em participantes randomizados para elafibranor comparado com placebo, mas não atingiu significância estatística (Tabela 2).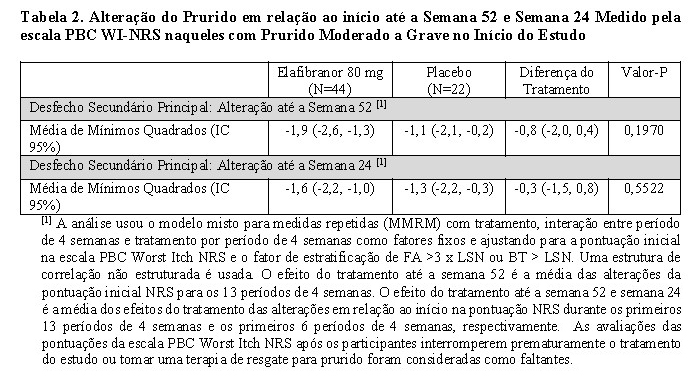
O tratamento com elafibranor foi associado com uma melhora no prurido como evidenciado por uma redução nas pontuações totais na escala de prurido PBC-40 e escala de prurido 5-D (EP5-D) comparada ao placebo na Semana 52 (Tabela 3).
Parâmetros lipídicos
Elafibranor demonstrou um efeito favorável nos parâmetros lipídicos. A redução média no colesterol de lipoproteína de baixa densidade (VLDL-C) e triglicérides (TG) foi maior nos participantes tratados com elafibranor comparado com placebo na Semana 52. A diferença média do LSM em relação ao placebo no VLDL-C foi -0,1 mmol/L [(IC95%: -0,2, -0,1); p < 0,001] e para TG foi -0,3 mmol/L [(IC95%: -0,4, -0,1)]; p < 0,001]. O colesterol de lipoproteína de alta densidade (HDL-C) permaneceu estável no tratamento com elafibranor.
Redução média no IgM
A redução média no IgM foi maior nos participantes tratados com elafibranor comparado com placebo na Semana 52. A diferença média do LSM em relação ao placebo no IgM foi -0,6 g/L [(IC95%: -0,9, -0,3); p < 0,001].
Redução média nos biomarcadores da síntese de ácido biliar
A redução média no C4 e no FGF-19 foi maior nos participantes tratados com elafibranor comparado com placebo na Semana 52. A diferença média do LSM em relação ao placebo no C4 foi -5,176 ug/L [(IC95%: -10,291; -0,062); p=0,0474]; e no FGF-19 foi -86,95 pg/ml [(IC95%: -170,37; -3,53); p=0,0412].
3. CARACTERÍSTICAS FARMACOLÓGICAS
Mecanismo de ação
Elafibranor e seu principal metabólito ativo GFT1007 são duplos agonistas dos receptores a?d ativados por proliferador de peroxissoma (PPAR).
A ativação do PPARa diminui a síntese do ácido biliar (AB), aumenta a desintoxicação do AB, e modula a saída do AB, resultando em uma diminuição da toxicidade da bile, e menos lesões aos colangiócitos e hepatócitos.
A ativação do PPARd também regula os transportadores que absorvem e secretam componentes da bile, contribuindo deste modo para diminuir a toxicidade da bile e melhorando a colestase.
A ativação do PPARa e PPARd também tem efeitos anti-inflamatórios em diferentes vias de inflamação, vias do fator nuclear kappa B (NF-kB) e B-cell lymphoma 6 (BCL6), respectivamente.
Efeitos farmacodinâmicos
No estudo pivotal de fase 3, ELATIVE, o tratamento com elafibranor resultou em uma redução marcante em relação à linha de base na fosfatase alcalina (FA) em apenas 4 semanas que foi mantida até a semana 52. Alinhado com a resposta bioquímica observada, com o tratamento com elafibranor, foram observadas reduções maiores nos biomarcadores da síntese de ácido biliar incluindo o precursor de ácido biliar 7 alfa-hidroxi-4-colesten-3-ona (C4) e Fator de Crescimento de Fibroblasto-19 (FGF-19), um regulador da síntese de ácido biliar. Em participantes tratados com elafibranor foram observadas diminuições significativas na Imunoglobulina M (IgM), Imunoglobulina G (IgG), e marcadores anti-inflamatórios comparadas ao placebo, alinhado com a demonstração in vitro das propriedades anti-inflamatórias do elafibranor.
Estudos in vitro em macrófagos, monócitos e células endoteliais humanas mostraram a capacidade do elafibranor e/ou GFT1007 de diminuir a secreção de marcadores inflamatórios tais como Proteína Quimioatraente de Monócitos-1 (MCP-1) e Interleucina-6 (IL-6) através de ativação combinada de PPARa e PPARd e mecanismos paralelos independentes de PPAR.
As propriedades antifibróticas do elafibranor foram demonstradas em células estreladas hepáticas primárias humanas (hHSCs), essenciais para a fibrogênese no fígado. Elafibranor inibe a proliferação de hHSC estimulada pelo Fator de Crescimento Derivado de Plaquetas (PDGF) de maneira dependente da dose por meio da modulação da fosforilação do PDGFRb. Além disso, elafibranor inibe a ativação de hHSC induzida pelo Fator de Crescimento Transformador Beta (TGFb1) no nível do gene, regulando negativamente, de maneira dose-dependente, a expressão de vários marcadores de fibrose, como alfa Actina do Músculo Liso (aSMA), Colágeno 1 alfa 1 (Col1a1) e Colágeno 4 alfa 1 (Col4a1), mas sem inibir a atividade quinase dos receptores TGFb1.
Eletrofisiologia cardíaca
A análise QT completa (TQT) excluiu qualquer efeito de prolongamento no intervalo QT/QTc causado pelo elafibranor em doses repetidas de até 300 mg por 14 dias.
Em estudos clínicos, não foram observadas alterações clinicamente significantes nos sinais vitais ou no eletrocardiograma (ECG) (incluindo intervalo QTc) em participantes tratados com elafibranor.
Propriedades farmacocinéticas
A exposição plasmática ao elafibranor (AUC) aumenta proporcionalmente de 50 a 360 mg (0,6 a 4,5 vezes à dose recomendada). O estado de equilíbrio é alcançado no dia 14 após doses diárias. A farmacocinética (PK) de elafibranor e seu principal metabólito ativo GFT1007 demonstrou ser independente do tempo após administração repetida por 16 dias. As exposições ao elafibranor e seu metabólito ativo nos participantes com CBP estão listadas na Tabela 4.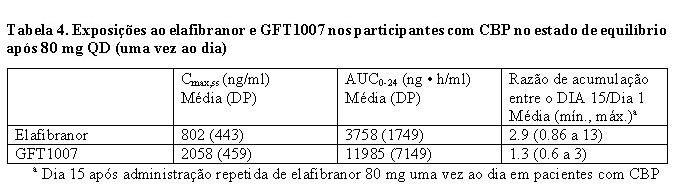
Absorção
Após administração oral repetida em participantes com CBP, o pico plasmático médio de elafibranor e GFT1007 em doses de 80 mg ocorreu dentro de 1,25 horas.
Quando administrado com uma refeição rica em gorduras e calorias, houve um atraso de 30 minutos no Tmax para elafibranor e um atraso de 1 hora para GFT1007 comparado com as condições de jejum. A Cmax e AUC plasmáticas do elafibranor diminuíram em 50% e 15% respectivamente e a Cmax plasmática do GFT1007 diminuiu em 30%, mas a AUC não foi afetada. Dados os níveis plasmáticos circulantes 2,25,3 vezes superiores do metabólito farmacologicamente ativo GFT1007 em comparação com elafibranor, a ingestão de alimentos tem impacto clínico limitado com base na exposição global do fármaco original e do seu metabólito ativo.
Distribuição
A ligação às proteínas plasmáticas do elafibranor e do GFT1007 é de aproximadamente 99,7% (principalmente à albumina sérica). O volume de distribuição aparente médio (Vd/F) do elafibranor em humanos é 4731L, após uma dose única de elafibranor de 80 mg em jejum.
Metabolismo
In vitro, elafibranor é metabolizado pela 15-cetoprostaglandina 13-? redutase (PTGR1). Estudo in vitro demonstraram que nem elafibranor nem GFT1007 apresentam metabolização importantes pelas principais isoformas do citocromo P450 (CYP) e uridina difosfato (UDP)-glucuronosiltransferase (UGT).
Após administração oral do elafibranor 14C radiomarcado, este foi rapidamente hidrolisado no metabólito ativo GFT1007. Dois metabólitos principais foram identificados no plasma, GFT1007 (metabólito ativo) e conjugados de glucuronídeo (metabólitos inativos).
Transportadores
Descobriu-se que in vitro elafibranor é um substrato para a proteína 2 associada à resistência a múltiplos fármacos dos transportadores intestinais (MRP2) e à proteína de resistência ao câncer de mama (BCRP). O papel do transporte de efluxo ativo é considerado insignificante em comparação com a absorção passiva, orientada por gradiente e de alta permeabilidade do elafibranor.
Eliminação
Após uma dose única de 80 mg em condições de jejum, a meia-vida de eliminação média é de 68,2 horas para elafibranor, e 15,4 horas para o metabólito GFT1007. O clearance aparente total médio de elafibranor (CL/F) foi 50,0 L/h após uma dose única de 80 mg em condições de jejum.
Excreção
Após uma dose única oral de 120 mg de elafibranor 14C radiomarcado em participantes saudáveis, aproximadamente 77,1% da dose foi recuperada nas fezes, principalmente como elafibranor (56,7% da dose administrada) e seu metabólito ativo GFT1007 (6,08% da dose administrada). Aproximadamente 19,3% da dose foi recuperada na urina, principalmente como conjugados de glucuronídeo.
Populações Especiais
Não houve evidência que idade (de 18 a 80 anos), gênero, raça, Índice de Massa Corporal (IMC) e estado renal, tenham qualquer impacto clinicamente significante sobre a farmacocinética do elafibranor e GFT1007.
Insuficiência Hepática
A exposição total ao fármaco e metabólito ativo não foi significantemente diferente entre os participantes com função hepática normal e com insuficiência hepática (Child Pugh A, B e C). Nenhum ajuste de dose é necessário para pacientes com insuficiência hepática leve (Child Pugh A) ou moderada (Child Pugh B). Entretanto, a fração não ligada do elafibranor e GFT1007 aumentou em aproximadamente 3 vezes em participantes com insuficiência hepática grave (Child Pugh C). Elafibranor não é recomendado para pacientes com insuficiência hepática grave (Child-Pugh C).
Interações Medicamentosas
Baseado em estudos in vitro, foi demonstrado que as enzimas CYP e UGT não desempenham um papel importante no metabolismo do elafibranor. Espera-se que as interações medicamentosas sejam mínimas com drogas que alteram significativamente a atividade do CYP ou UGT.
Etinilestradiol/levonorgestrel (substratos do CYP3A4)
A Cmax de etinilestradiol diminuiu 20% e a AUCinf diminuiu 35% após o uso concomitante de uma dose única de contraceptivo oral combinado (0,02 mg de etinilestradiol e 0,15 mg de levonorgestrel) e elafibranor 80 mg uma vez ao dia em estado de equilíbrio, enquanto a farmacocinética do levonorgestrel permaneceu inalterada.
Estudos Clínicos
Varfarina (substrato CYP2C9): Administração concomitante de elafibranor com varfarina não resultou em aumento na exposição (AUC, Cmax) de varfarina, e nenhuma diferença na razão normalizada internacional (RNI) comparado à varfarina sozinha.
Sinvastatina e atorvastatina (substratos do CYP3A, polipeptídios transportadores de ânions orgânicos 1B1 (OATP1B1) e OATP1B3): A administração concomitante de doses repetidas de elafibranor com sinvastatina, ou atorvastatina, não resultou em alteração clinicamente significativa na exposição da sinvastatina ou seu metabólito b-Hidroxiácido, ou atorvastatina.
A Cmax e a AUCinf da atorvastatina diminuíram 28% e 11%, respectivamente, após o uso concomitante de uma dose única de atorvastatina 40 mg e elafibranor 180 mg uma vez ao dia em estado de equilíbrio.
A Cmax e a AUCinf do metabólito ativo da sinvastatina, o ácido b-hidroxídico da sinvastatina, diminuíram 26% e 32%, respectivamente, após o uso concomitante de uma dose única de sinvastatina 20 mg e elafibranor 80 mg uma vez ao dia em estado de equilíbrio.
Indometacina (inibidor do PTGR1): Após estudos clínicos de interação medicamentosa, não foi observado efeito na PK clínica do elafibranor com a coadministração de indometacina.
Sitagliptina (inibidor da dipeptidil peptidase-IV (DPP-IV)): Não foram observados efeitos clinicamente significantes quando se coadministrou elafibranor com a sitagliptina.
Estudos In vitro
Inibição e indução do citocromo P450 (CYP): Elafibranor e GFT1007 não foram considerados inibidores de CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 e CYP3A4. Não foi observada inibição tempo-dependente do CYP.
Elafibranor e GFT1007 não causaram indução do CYP1A2, CYP2B6 e CYP3A4.
Inibição do UGT: Baseado em dados in vitro não se espera que elafibranor iniba o UGT1A1, 1A3, 1A4, 1A6, 1A9, 2B7, 2B10 e 2B15.
GFT1007 inibiu UGT1A6, mas não UGT1A1, 1A3, 1A4, 1A9, 2B7, 2B10 e 2B15.
Sistemas Transportadores: Elafibranor foi um inibidor de OATP1B3 e BCRP, mas não um inibidor da glicoproteína de permeabilidade/proteína de resistência a múltiplas drogas 1 (P-gp/MDR1), OATP1B1, transportador de cátions orgânicos 1 (OCT1), OCT2, transportador de ânions orgânicos 1 (OAT1), proteína de extrusão de múltiplas drogas e toxinas 1 (MATE1), MATE2-K, OAT3 e bomba de exportação de sais biliares (BSEP).
GFT1007 não foi considerado um inibidor de OAT3, OATP1B3, BSEP, P-gp/MDR1, BCRP, OATP1B1, OCT1, OCT2, OAT1, MATE1 e MATE2-K.
Dados de segurança pré-clínica
Os dados não-clínicos não revelam riscos especiais para o ser humano, segundo estudos convencionais de farmacologia de segurança, toxicidade de dose repetida, genotoxicidade e potencial carcinogênico.
Carcinogênese/mutagênese
Elafibranor foi avaliado em dois estudos de carcinogenicidade em camundongos e ratos, com administração oral por gavagem por até dois anos na dose de 1, 3, 10, ou 30 mg/kg/dia. Até a dose mais elevada testada em ambas as espécies (aproximadamente 3 e 6 vezes a exposição da AUC na dose humana máxima recomendada (DHMR) de 80 mg/dia), a administração de elafibranor não resultou em um aumento da incidência de tumores que sejam relevantes para humanos.
Elafibranor, seu principal metabólito ativo GFT1007 e o metabólito racêmico de acil glucuronídeo GFT3351 foram desprovidos de potencial genotóxico em uma bateria abrangente de ensaios de genotoxicidade in vivo e/ou in vitro.
Toxicidade e farmacologia animal
A toxicologia geral foi avaliada em espécies de roedores (camundongos e ratos) e não-roedores (macacos) após administração oral de dose única e repetida por até 6 meses em ratos e 12 meses em macacos, respectivamente.
Elafibranor exibiu um perfil de segurança favorável quando administrado em doses orais únicas em estudos de toxicidade aguda em ratos e camundongos. A administração oral de doses repetidas por até 6 meses em ratos e 12 meses em macacos não revelou qualquer sinal de toxicidade relevante para humanos até a dose máxima testada de 100 e 50 mg/kg/dia, respectivamente (correspondendo a ~15 vezes a exposição AUC na DHMR em estudos com ratos e macacos).
Além disso, até em doses mais elevadas testadas, elafibranor não demonstrou quaisquer efeitos relevantes nos órgãos e sistemas anteriormente descritos como sendo uma preocupação de segurança com os agonistas do PPARc (aumento de peso, hemodiluição, retenção de líquidos que conduz a insuficiência cardíaca congestiva, câncer de bexiga).
Toxicidade de reprodução e desenvolvimento
Elafibranor não tem efeitos adversos na fertilidade de ratos ou no desenvolvimento embrionário inicial até 100 mg/kg/dia (correspondendo a aproximadamente 17 vezes a exposição AUC na DHMR).
Elafibranor não mostrou evidências de toxicidade no desenvolvimento de ratos e coelhos.
Em ratas prenhas, a administração oral de elafibranor uma vez ao dia durante o período de organogênese do dia de gestação (DG) 6 ao DG17 não resultou em efeitos sobre o desenvolvimento embriofetal na dose de até 300 mg/kg/dia (~100 vezes a exposição AUC na DHMR). No entanto, no estudo de desenvolvimento pré e pós-natal em ratos, a administração oral de elafibranor uma vez ao dia durante a gravidez e a lactação (do DG6 ao dia de lactação (DL)21) foi associada à redução da sobrevivência dos filhotes (especialmente, mas não exclusivamente, no período perinatal inicial), descoloração azul/preta da seção da cauda de alguns filhotes, menor peso corporal dos filhotes e atrasos no desenvolvimento em todas as doses (de exposições maternas de 2 vezes a exposição AUC na DHMR e acima). Com exposições maternas iguais ou superiores a 4,5 vezes a exposição AUC na DHMR, houve evidência de trombose arterial aórtica ou ilíaca em filhotes falecidos prematuramente. Em filhotes coletados no dia 14 pós-natal, o elafibranor não foi detectado e apenas a exposição plasmática mínima ao seu metabólito ativo foi detectável em filhotes do grupo de dose mais alta, onde as exposições maternas foram 16 vezes acima da exposição AUC na DHMR. A prole adulta sobrevivente não apresentou efeitos do elafibranor no aprendizado e na memória, no desenvolvimento reflexo ou na capacidade reprodutiva.
Em coelhas prenhas, a administração de elafibranor durante a organogênese na dose elevada de 300 mg/kg/dia (3 vezes a exposição AUC na DHMR), foi associada à toxicidade materna acentuada, aumento da letalidade embrionária, redução do peso fetal e uma baixa incidência de malformações fetais. Na dose média de 100 mg/kg/dia (0,5 vezes a exposição AUC na DHMR), apesar da toxicidade materna, não houve efeito na sobrevivência, no peso fetal, nem nas malformações fetais. O único achado foram variações de ossificação fetal nos ossos distais dos membros. Não foram observados efeitos adversos no desenvolvimento embriofetal com a dose baixa de 30 mg/kg/dia (ou seja, aproximadamente 0,1 vezes a exposição AUC na DHMR).
Farmacologia de segurança
Não foram identificados problemas de segurança ao avaliar os efeitos potenciais do elafibranor nos sistemas cardiovascular, respiratório e nervoso central.
Potencial fototóxico
No ensaio in vitro utilizando fibroblastos 3T3, elafibranor, mas não o GFT1007, apresentou potencial fototóxico. A partir do estudo de acompanhamento UV-LLNA em camundongos, conclui-se que não há risco de fototoxicidade in vivo associado com elafibranor até a dose mais alta testada de 800 mg/kg/dia (~46 vezes a exposição Cmax na DHMR). Além disso, estudos de distribuição nos tecidos utilizando elafibranor radiomarcado em camundongos, ratos e macacos demonstraram que não houve acumulação de elafibranor nos olhos e na pele, o que limita o seu potencial fototóxico.
4. CONTRAINDICAÇÕES
IQRVO® é contraindicado a pessoas com hipersensibilidade à substância ativa ou a qualquer um dos excipientes da formulação.
5. ADVERTÊNCIAS E PRECAUÇÕES
Eventos relacionados ao fígado
Aumentos nos testes bioquímicos hepáticos incluindo aumento nas transaminases e bilirrubina foram reportados em 3,7% dos participantes recebendo elafibranor comparado a 5,7% dos participantes recebendo placebo.
Avaliações clínicas e laboratoriais da função hepática devem ser feitas antes do início e após o tratamento, de acordo com o gerenciamento de rotina do paciente.
Se forem observados aumentos nos testes bioquímicos hepáticos e/ou disfunção hepática, é recomendada uma imediata investigação da causa e a interrupção do tratamento com elafibranor deve ser considerada.
Insuficiência hepática
IQIRVO® não é recomendado para pacientes que possuem ou desenvolvem cirrose descompensada (por exemplo, ascite, sangramento por varizes, encefalopatia hepática).
Creatina fosfoquinase sérica elevada e lesão muscular
Aumentos na creatina fosfoquinase (CPK) sérica foram relatados em participantes recebendo elafibranor (3,7% no grupo elafibranor comparado com 0% no grupo placebo). Além dos relatos de aumento de CPK, houve um caso de rabdomiólise que ocorreu no estudo pivotal fase 3, ELATIVE, em um participante com cirrose e em tratamento com um inibidor da HMG-CoA redutase. CPK deve ser avaliada antes do início do tratamento com elafibranor e depois disso, de acordo com o gerenciamento de rotina do paciente. Medidas periódicas de CPK podem ser consideradas em pacientes iniciando o tratamento com elafibranor, especialmente aqueles com inibidores da HMG-CoA redutase concomitantes. Pacientes em tratamento com elafibranor devem ser aconselhados a relatar ao seu médico quaisquer sintomas musculares inexplicáveis, tais como dor e fraqueza. Se forem observados aumentos na CPK ou sinais e sintomas inexplicados de lesão muscular, é recomendada uma investigação da causa imediata e a interrupção do tratamento com elafibranor deve ser considerada. Vide item 9. REAÇÕES ADVERSAS.
Toxicidade embrio-fetal
Baseado em dados de estudos em animais, elafibranor pode causar dano fetal quando administrado a mulheres grávidas. Pacientes devem ser informados do risco potencial para o feto se elafibranor for administrado durante a gravidez.
Fertilidade, gravidez e lactação
Fertilidade
Não há dados humanos disponíveis sobre o efeito de elafibranor na fertilidade. Estudos em animais não indicam quaisquer efeitos diretos ou indiretos na fertilidade ou na habilidade para reproduzir.
Mulheres com potencial para engravidar/contracepção
Devido ao dano potencial ao feto, elafibranor não é recomendado para mulheres com potencial para engravidar que não estejam usando método contraceptivo efetivo.
O elafibranor pode reduzir a eficácia de contraceptivos que contenham etinilestradiol e, portanto, mulheres com potencial para engravidar devem usar métodos contraceptivos não hormonais eficazes ou adicionar um método de barreira quando utilizarem contraceptivos hormonais,
Mulheres com potencial para engravidar devem continuar a usar método efetivo de contracepção por 3 semanas após a dose final de elafibranor. Antes de iniciar o tratamento com elafibranor em pacientes com potencial de engravidar, deve-se verificar se não estão grávidas.
Recomenda-se que mulheres que estejam planejando engravidar busquem aconselhamento com seu médico sobre opções alternativas de tratamento.
Gravidez
Há dados limitados sobre o uso de elafibranor em mulheres grávidas.
Estudos em animais prenhas com elafibranor indicam efeitos adversos (perda fetal, malformações, natimortos e/ou mortes perinatais) em exposição clinicamente relevante.
Elafibranor não é recomendado durante a gravidez por causa do dano potencial ao feto.
Categoria de risco C: Este medicamento não deve ser usado por mulheres grávidas sem orientação de um médico ou cirurgião-dentista.
Lactação
Não se sabe se elafibranor ou seus metabólitos são excretados no leite humano. Quando elafibranor foi administrado a ratas durante a gravidez e lactação, houve redução da sobrevivência e do crescimento da prole em exposições maternas próximas das exposições ao paciente e não está claro se a excreção de elafibranor ou do seu metabólito no leite contribuiu para os efeitos adversos na prole.
Elafibranor não é recomendado durante a lactação e por pelo menos 3 semanas após a última dose de elafibranor porque o risco ao lactente não pode ser excluído.
Efeitos sobre a capacidade de dirigir e usar máquinas
Elafibranor não tem influência sobre a capacidade de dirigir e usar máquinas.
Atenção: Contém os corantes dióxido de titânio, óxido de ferro amarelo e óxido de ferro vermelho.
6. INTERAÇÕES MEDICAMENTOSAS
Com base nos estudos clínicos realizados, o elafibranor pode ser um indutor leve do CYP3A4.
Um estudo de interação clínica demonstrou que a coadministração de elafibranor e um contraceptivo hormonal oral combinado produziu uma redução de etinilestradiol, o que pode levar a falha contraceptiva e/ou aumento de sangramento de escape, enquanto a exposição ao levonorgestrel permaneceu inalterada.
Sequestrantes de ligação de ácidos biliares: Os sequestrantes de ácidos biliares podem interferir na ação do IQIRVO® reduzindo sua absorção e exposição sistêmica, o que pode reduzir a eficácia do IQIRVO®. Administrar IQIRVO® pelo menos quatro horas antes ou quatro horas depois de tomar um sequestrante de ligação de ácido biliar, ou no maior intervalo possível.
7. CUIDADOS DE ARMAZENAMENTO DO MEDICAMENTO
IQIRVO® deve ser mantido em temperatura ambiente (entre 15 e 30°C).
IQIRVO® tem um prazo de validade de 36 meses a partir da data de fabricação.
Número de lote e datas de fabricação e validade: vide embalagem.
Não use medicamento com o prazo de validade vencido. Guarde-o em sua embalagem original.
Após a primeira abertura do frasco, o produto pode ser armazenado por, no máximo, 30 dias.
IQIRVO® é um comprimido revestido, redondo, laranja, de aproximadamente 8 mm de diâmetro, gravado com 'ELA 80' em um dos lados.
Antes de usar, observe o aspecto do medicamento.
Todo medicamento deve ser mantido fora do alcance das crianças.
8. POSOLOGIA E MODO DE USAR
IQIRVO® é um medicamento de uso exclusivamente oral.
A dose recomendada é 80 mg uma vez ao dia com ou sem alimento.
Antes de iniciar o tratamento com IQIRVO®:
- Verifique se as mulheres com potencial reprodutivo não estejam grávidas antes de iniciar o tratamento com IQIRVO®.
Modificação de Administração para Sequestrantes de Ácido Biliar:
Administrar IQIRVO® pelo menos 4 horas antes ou 4 horas depois do uso de sequestrante de ácido biliar.
Populações Especiais
Pacientes idosos (65 anos de idade e acima)
Não é necessário ajuste de dose em pacientes acima de 65 anos de idade.
Insuficiência renal
Não é necessário ajuste de dose em pacientes com insuficiência renal.
Insuficiência hepática
Não é necessário ajuste de dose em pacientes com insuficiência hepática leve (Child-Pugh A) ou moderada (Child-Pugh B).
A segurança e eficácia de elafibranor não foram estabelecidas em pacientes com colangite biliar primária com insuficiência hepática grave. Não é recomendado o uso em pacientes com insuficiência hepática grave (Child-Pugh C).
A segurança e eficácia de IQIRVO® em pacientes com cirrose descompensada não foram estabelecidas. O uso de IQIRVO® não é recomendado em pacientes que têm ou desenvolvem cirrose descompensada (por exemplo, ascite, sangramento varicoso, encefalopatia hepática). Monitore pacientes com cirrose para evidências de descompensação. Considere interromper IQIRVO® se o paciente progredir para insuficiência hepática moderada ou grave (Child-Pugh B ou C).
População pediátrica
O medicamento IQIRVO® não foi estudado na população pediátrica (abaixo de 18 anos), então não é recomendado o uso nesta população.
Dose esquecida
Em caso de esquecimento de uma dose de elafibranor, o paciente não deve tomar a dose esquecida e sim tomar a dose subsequente no próximo horário estipulado. O paciente não deve dobrar a dose para compensar a dose perdida.
Este medicamento não deve ser partido ou mastigado.
9. REAÇÕES ADVERSAS
Resumo do perfil de segurança
No estudo pivotal fase 3, ELATIVE, 161 participantes foram randomizados numa razão 2:1 para receber elafibranor 80 mg (n=108) ou placebo (n=53) por pelo menos 52 semanas. Ao final do período duplo-cego do estudo, a duração mediana da exposição foi de 63,07 e 61,00 semanas nos grupos elafibranor e placebo, respectivamente.
As reações adversas mais comumente reportadas associadas com elafibranor em mais de 10% dos participantes (n=108) foram dor abdominal (11,1%), diarreia (11,1%), náusea (11,1%) e vômito (11,1%). Estas reações não foram graves e eram de intensidade leve a moderada, ocorridas no início do tratamento e geralmente se resolveram em poucos dias a algumas semanas, sem qualquer modificação de dose ou medidas de suporte.
A reação adversa à droga mais comum levando à descontinuação do tratamento foi aumento da CPK sérica (3,7%).
Lista tabulada de reações adversas
Dentro da classe de Sistema de órgãos, as reações adversas são listadas por frequência usando as seguintes categorias: muito comum (≥1/10), comum (≥1/100 a < 1/100), incomum (≥1/1 000 a < 1/100), rara (≥1/10 000 a < 1/1 000), muito rara ( < 1/10 000), desconhecido (não pode ser estimada a partir dos dados disponíveis).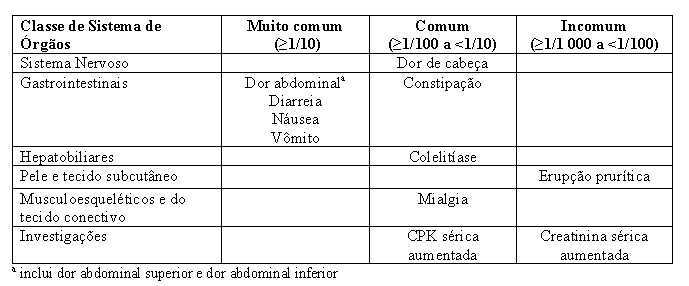
Descrição de reações adversas selecionadas
Nove (8,3%) participantes no grupo elafibranor e 6 (11,3%) participantes no grupo placebo tiveram dor de cabeça. Entretanto, dentro dos primeiros 10 dias do tratamento do estudo, mais participantes no grupo elafibranor tiveram dor de cabeça comparado com o grupo placebo (3,7% comparado a 0% respectivamente).
Quatro (3,7%) participantes no grupo elafibranor e nenhum participante no grupo placebo tiveram aumento significante de CPK sérica, levando à descontinuação da droga. Em 2 dos 4 participantes, a CPK estava > 5 x o limite superior da normalidade (LSN). Todos os eventos foram não-graves e de intensidade leve a moderada. Dois dos participantes também experimentaram sintomas associados a mialgia. No início do estudo, os valores médios de CPK eram similares entre os grupos de tratamento e dentro da faixa de normalidade; os valores na Semana 52 permaneceram dentro da faixa de normalidade em ambos os grupos. A alteração média em relação ao início na Semana 52 foi 6,2 (38,1) U/L no grupo elafibranor e 12,3 (67,0) U/L no grupo placebo.
Atenção: este produto é um medicamento novo e, embora as pesquisas tenham indicado eficácia e segurança aceitáveis, mesmo que indicado e utilizado corretamente, podem ocorrer eventos adversos imprevisíveis ou desconhecidos. Nesse caso, notifique os eventos adversos pelo Sistema VigiMed, disponível no Portal da Anvisa.
10.SUPERDOSE
No caso de suspeita de superdose, os pacientes devem ser cuidadosamente observados, e tratamento sintomático apropriado e cuidados de suporte devem ser iniciados.
Em caso de intoxicação ligue para 0800 722 6001, se você precisar de mais orientações.
REFERÊNCIAS BIBLIOGRÁFICAS
-Schattenberg JM, Pares A, Kowdley KV, Heneghan MA, Caldwell S, Pratt D, Bonder A, HirschfieldGM, Levy C, Vierling J, Jones D, Tailleux A, Staels B, Megnien S, Hanf R, Magrez D, Birman P,Luketic V. A randomized placebo-controlled trial of elafibranor in patients with primary biliarycholangitis and incomplete response to UDCA. J Hepatol. 2021 Jun;74(6):1344-1354. doi:10.1016/j.jhep.2021.01.013. Epub 2021 Jan 21. PMID: 33484775.
-Investigator's Brochure Elafibranor (GFT505) Primary Biliary Cholangitis (PBC) and PrimarySclerosing Cholangitis (PSC) version no 27, dated on 23 September 2022, (iDocS ID 524429)
-Integrated Safety Summary (ISS): ISS Tables (iDocS ID 485931), ISS Listings (iDocS ID 671618)
-Final CSR pivotal ELATIVE phase 3 study V1.0 dated on 31 Aug2023 (iDocS ID 557679)
-Ocaliva 5 mg, 10 mg film-coated tablets (obeticholic acid) EU SmPC revision date 09 November2022
-Ocaliva 5 mg, 10 mg film-coated tablets (obeticholic acid) US PI revision date 05/2022
-Aaron Wetten, David Emrys Jeffreys Jones & Jessica Katharine Dyson (2022) Seladelpar: aninvestigational drug for the treatment of early-stage primary biliary cholangitis (PBC), ExpertOpinion on Investigational Drugs, 31:10, 1101-1107, DOI:10.1080/13543784.2022.2130750
CTD modules 2.4 (iDocS ID 601147), 2.5 (iDocS ID 485932), 2.7.3 (iDocS 485935), 2.7.4 (iDocS ID 485936)
-SER signed on 6 OCT 2025 (iDocs ID: 860507)
-CSR CLIN-60190-468 (iDocs ID: 798611)
-Addendum Clinical Overview Module 2.5 (iDocs ID: 861113)
-Study GFT505B-319-1 CSR Listing 16.2.7.1: (ID: 557679)
-Section 11.2.6.1 of the CSR of clinical study GFT505B-319-1 (ID: 557679)
-Cmax and AUC Mean (SD) Study 216-1 CSR (ID: 781520), page 132
-CSR GFT505-109-5 study, section 11.4 (ID: 781459)
-CSR GFT505-115-11 study, Table 11.1.2-1, Table 11.1.2-2, Table 11.1.3-1 (ID: 619404)
Dizeres legais.
Registro: 1.6977.0006
VENDA SOB PRESCRIÇÃO
Esta bula foi aprovada pela Anvisa em 07/04/2026.