PADCEV
PFIZER
enfortumabe vedotina
Antineoplásico.
Apresentações.
Padcev® pó liofilizado para solução injetável contendo 20 mg ou 30 mg de enfortumabe vedotina em embalagens com 1 frasco-ampola de dose única.
VIA DE ADMINISTRAÇÃO: VIA INTRAVENOSA
USO ADULTO
Composição.
Cada frasco-ampola de dose única de Padcev® contém 20 mg ou 30 mg de enfortumabe vedotina. Após reconstituição, cada mL contém 10 mg de enfortumabe vedotina. Excipientes: histidina, cloridrato de histidina monoidratado, trealose di-hidratada e polissorbato 20.
Informações técnicas.
1. INDICAÇÕES
Padcev®, em associação com pembrolizumabe, é indicado para o tratamento de pacientes adultos com carcinoma urotelial localmente avançado ou metastático (mUC). Padcev® como agente único, é indicado para o tratamento de pacientes adultos com carcinoma urotelial localmente avançado ou metastático (mUC) que:
• Tenham recebido previamente um inibidor do receptor 1 de morte programada (PD-1) ou ligante 1 de
morte programada (PD-L1) e quimioterapia contendo platina, ou
• São inelegíveis para quimioterapia contendo cisplatina e tenham recebido previamente uma ou mais
linhas de tratamento prévias.
2. RESULTADOS DE EFICÁCIA
Estudos clínicos
Carcinoma urotelial localmente avançado ou metastático previamente tratado
Estudo clínico EV-3011
A eficácia de Padcev® como agente único foi avaliada no estudo EV-301, um estudo aberto, randomizado e multicêntrico que incluiu 608 pacientes com carcinoma urotelial localmente avançado ou metastático que receberam tratamento prévio com um inibidor de PD-1 ou PD-L1 e uma quimioterapia baseada em platina. Os pacientes foram randomizados em 1:1 para receber Padcev® na dose de 1,25 mg/kg nos Dias 1, 8 e 15 de um ciclo de 28 dias ou a quimioterapia de escolha do investigador. A randomização foi estratificada por Eastern Cooperative Oncology Group (ECOG OS) (0 vs 1), região do mundo (Europa Ocidental vs EUA vs demais países) e presença de metástase hepática.
Os pacientes foram excluídos do estudo se apresentassem metástases ativas ao sistema nervoso central (SNC), neuropatia sensorial ou motora em andamento Grau ≥2 ou diabetes não controlado definido como hemoglobina A1C (HbA1c) ≥8% ou HbA1c ≥7% com sintomas de diabetes associados.
A idade mediana foi de 68 anos (variação: 30 a 88 anos) e 77% eram do sexo masculino. Os dados demográficos raciais foram relatados como caucasianos (52%), asiáticos (33%), negros (0,7%), nativos do Havaí ou de outra ilha do Pacífico (0,2%) ou raça não relatada (15%). Nove por cento dos pacientes eram hispânicos ou latinos. Todos os pacientes apresentaram um estado de desempenho ECOG de 0 (40%) ou 1 (60%). Trinta e quatro por cento dos pacientes tinham tumores localizados no trato superior, que incluíam a pelve renal e ureter. Oitenta por cento dos pacientes apresentaram metástases viscerais incluindo 31% com metástases hepáticas. Setenta e seis por cento dos pacientes apresentava histologia de carcinoma de células transicionais (TCC) puro; 14% apresentavam TCC com outras variantes histológicas; e 10% apresentavam outras histologias tumorais, incluindo adenocarcinoma e carcinoma de células escamosas. O número mediano de terapias prévias foi 2 (intervalo 1 a ≥3). Sessenta e três por cento dos pacientes receberam esquemas anteriores à base de cisplatina, 26% receberam esquemas anteriores à base de carboplatina, e outros 11% receberam esquemas à base de cisplatina e carboplatina. Os pacientes no braço de controle receberam docetaxel (38%), paclitaxel (36%) ou vinflunina (25%).
As principais medidas de resultados de eficácia foram a sobrevida global (SG), a sobrevida livre de progressão (SLP) e a taxa de resposta global (ORR) avaliadas pelo investigador usando RECIST v1.1. Os resultados de eficácia foram consistentes em todos os subgrupos estratificados de pacientes.
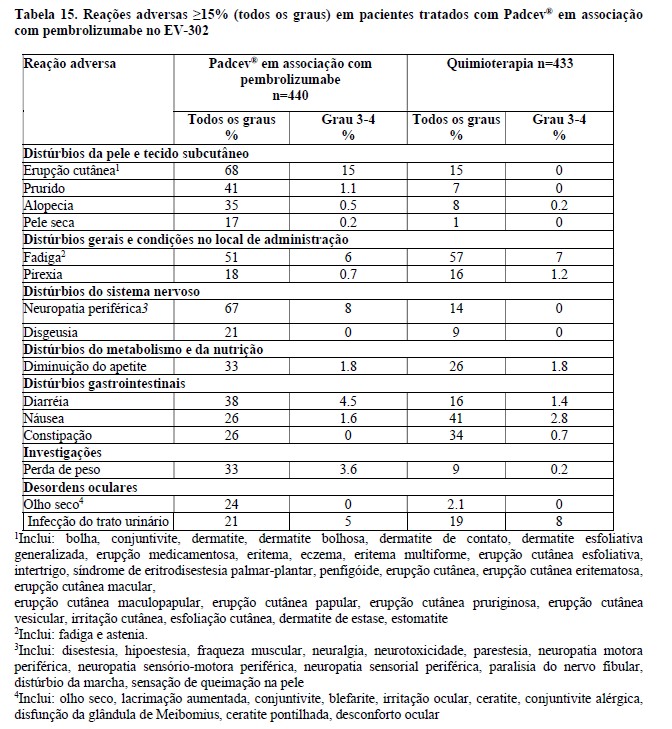
A tabela 1 e as figuras 1 e 2 resumem os resultados de eficácia para EV-301.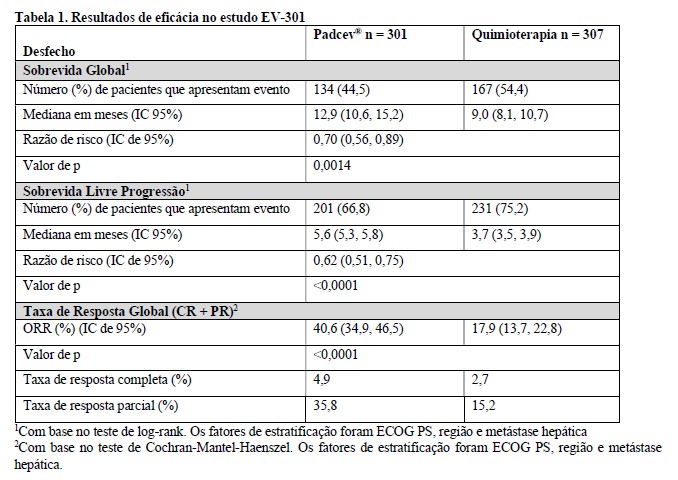
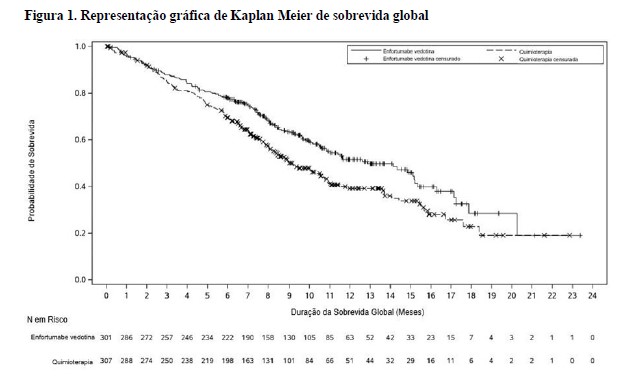
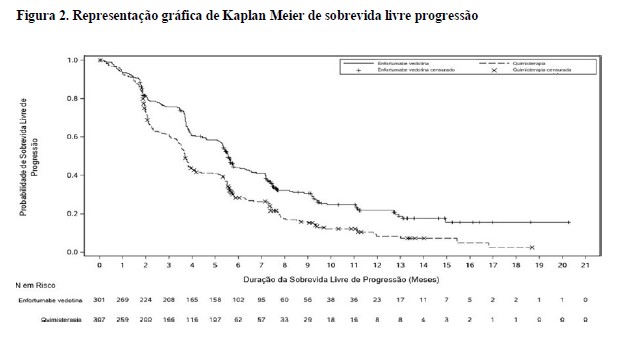
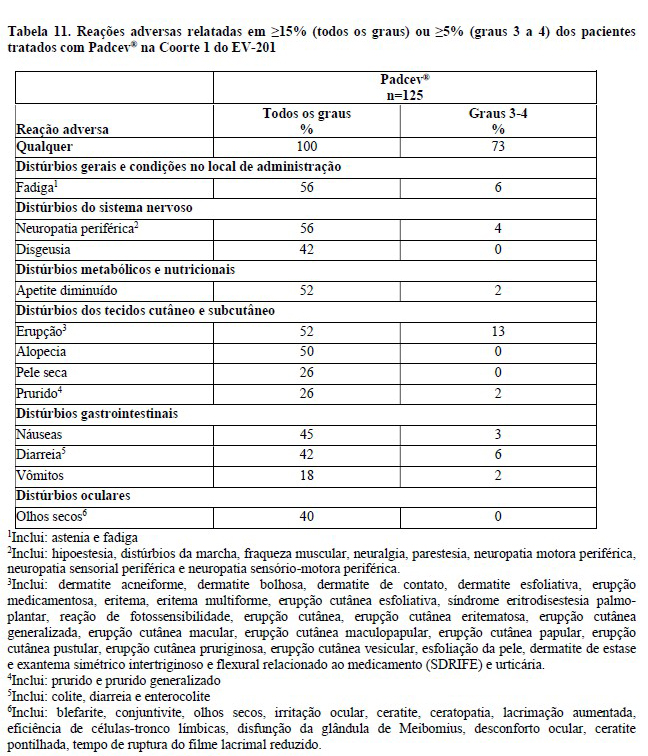
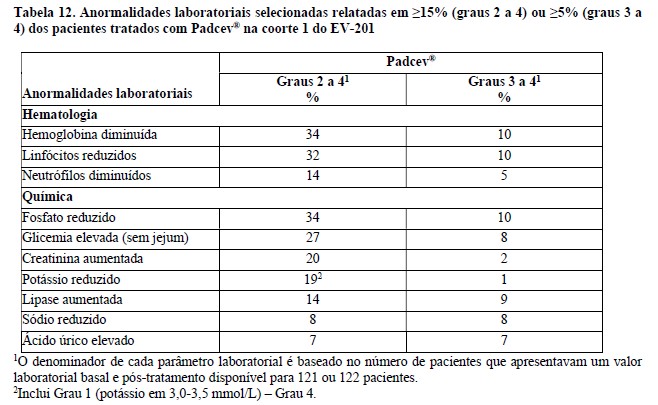
Estudo clínico EV-201, Coorte 12
A eficácia de Padcev® como agente único também foi investigada na Coorte 1 do estudo EV-201, um estudo de braço único, de coortes múltiplas e multicêntrico que incluiu 125 pacientes com carcinoma urotelial localmente avançado ou metastático que receberam tratamento prévio com um inibidor de PD-1 ou PD-L1 e uma quimioterapia baseada em platina. Os pacientes foram excluídos do estudo se apresentassem metástases ativas ao sistema nervoso central (SNC), neuropatia sensorial ou motora em andamento Grau ≥2, insuficiência cardíaca ou diabetes não controlado definido como hemoglobina A1C (HbA1c) ≥8% ou HbA1c ≥7% com sintomas de diabetes associados.
Padcev® foi administrado na dose de 1,25 mg/kg, como uma infusão via intravenosa (IV) nos dias 1, 8, e 15 de cada ciclo de 28 dias.
A idade mediana foi de 69 anos (variação: 40 a 84 anos) e 70% eram do sexo masculino. Os dados demográficos raciais foram relatados como caucasianos (85%), asiáticos (9%), negros (2%), outro (0,8%) ou raça não relatada (4%). Quatro por cento dos pacientes eram hispânicos ou latinos. Todos os pacientes apresentaram um estado de desempenho do ECOG de 0 (32%) ou 1 (68%). Noventa por cento dos pacientes apresentaram metástases viscerais incluindo 40% com metástases hepáticas. Aproximadamente dois terços (67%) dos pacientes apresentaram histologia de carcinoma de células transicionais (TCC) puro; 33% apresentaram TCC com outras variantes histológicas. O número mediano de terapias sistêmicas prévias foi 3 (variação: 1 a 6). Sessenta e seis por cento dos pacientes receberam esquemas prévios à base de cisplatina, 26% receberam esquemas anteriores à base de carboplatina, e outros 8% receberam esquemas à base de cisplatina e carboplatina.
As principais medidas de desfecho de eficácia foram a taxa de resposta objetiva confirmada (ORR) e a duração da resposta (DOR) avaliadas por revisão central independente em caráter cego (blinded independent central review: BICR) utilizando RECIST v1.1.
Os resultados de eficácia são apresentados na Tabela 2.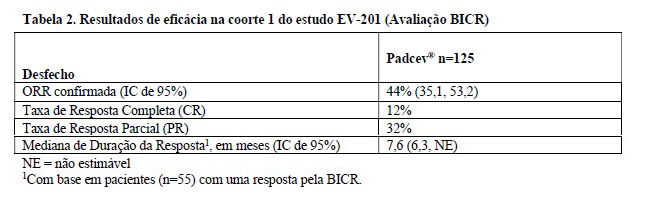
Pacientes inelegíveis à cisplatina com carcinoma urotelial localmente avançado ou metastático
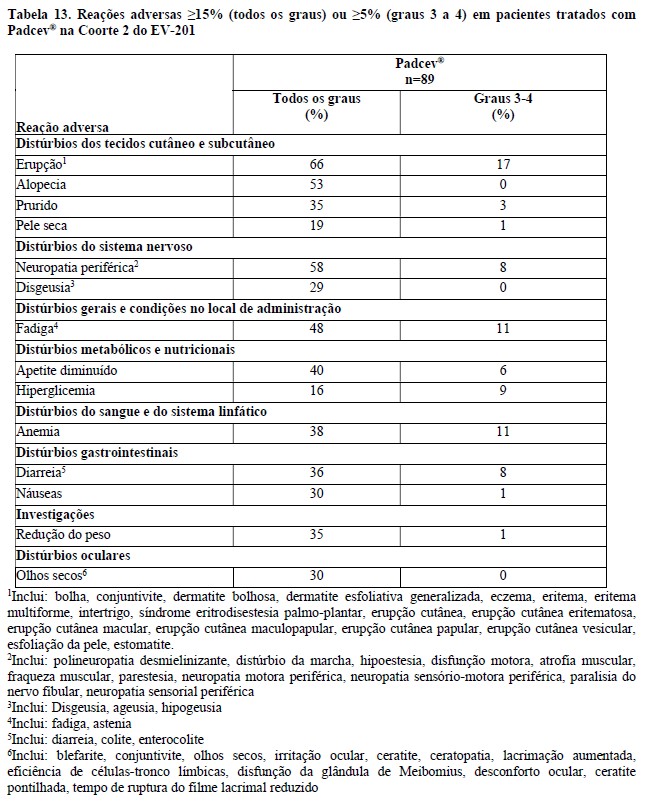
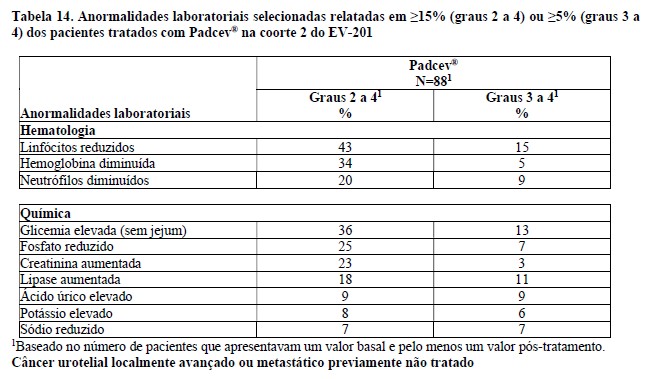
Estudo clínico EV-201, Coorte 22
A eficácia de Padcev® como agente único também foi avaliada na Coorte 2 do EV-201, um estudo multicentrico, de braço único, multicoorte, em 89 pacientes com carcinoma urotelial localmente avançado ou metastático que receberam tratamento prévio com um inibidor de PD-1 ou PD-L1 e eram inelegíveis para cisplatina e que não receberam platina no cenário localmente avançado ou metastático. Os pacientes foram excluídos do estudo se apresentassem metástases ativas ao SNC, neuropatia sensorial ou motora em andamento Grau ≥2, insuficiência cardíaca ou diabetes não controlado definido como hemoglobina A1C (HbA1c) ≥8% ou HbA1c ≥7% com sintomas de diabetes associados.
Padcev® foi administrado na dose de 1,25 mg/kg, como uma infusão via intravenosa (IV) nos dias 1, 8, e 15 de cada ciclo de 28 dias.
A idade mediana foi de 75 anos (variação: 49 a 90 anos) e 74% eram do sexo masculino. Os dados demográficos raciais foram relatados como caucasianos (70%), asiáticos (22%) ou raça não relatada (8%). Um por cento dos pacientes eram hispânicos ou latinos. Os pacientes tiveram um status de desempenho do ECOG basal de 0 (42%), 1 (46%) e 2 (12%). Quarenta e três por cento dos pacientes apresentavam tumores localizados no trato superior, que incluíam a pelve renal e ureter. Setenta e nove por cento dos pacientes apresentaram metástases viscerais e 24% apresentaram metástases hepáticas. As razões para inelegibilidade para cisplatina incluíam: 66% com creatinina basal 30 a 59 mL/min, 7% com ECOG PS de 2, 15% com perda de audição de grau 2 ou superior e 12% com mais de um critério de inelegibilidade para cisplatina. Setenta por cento dos pacientes apresentaram histologia de TCC; 13% apresentaram TCC com diferenciação escamosa e 17% apresentaram TCC com outras variantes histológicas. O número mediano de terapias sistêmicas prévias foi 1 (variação: 1 a 4). Os resultados de eficácia são apresentados na Tabela 3 abaixo.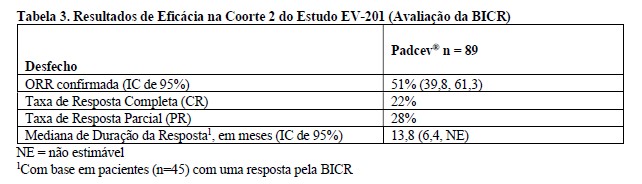
Pacientes inelegíveis para cisplatina com carcinoma urotelial localmente avançado ou metastático não tratados anteriormente
Estudo Clínico EV-1033
A combinação de Padcev® com pembrolizumabe foi investigada no estudo EV-103, um estudo aberto de Fase lb/2 de múltiplas coortes (coorte de escalonamento de dose, Coorte A, Coorte K) em pacientes com câncer urotelial localmente avançado ou metastático que eram inelegíveis para quimioterapia contendo cisplatina e não receberam terapia sistêmica anterior para doença localmente avançada ou metastática. O estudo excluiu pacientes com doença autoimune ou condição médica que requeresse imunossupressão, metástases ativas no sistema nervoso central (SNC), neuropatia sensorial ou motora contínua Grau 2 ou diabetes descompensada definida como hemoglobina AlC (HbAlc) 8% ou HbAlc 2: 7% com sintomas de diabetes associados.
A inelegibilidade à cisplatina para pacientes na coorte de escalonamento de dose foi determinada pelo investigador. Pacientes na coorte A foram considerados inelegíveis à cisplatina se eles tivessem pelo menos um dos seguintes critérios: depuração de creatinina basal 30-59 ml/min, ECOG PS 2, perda/disfunção auditiva, idade ou alergia à cisplatina. Pacientes na coorte K foram considerados inelegíveis à cisplatina se eles tivessem pelo menos um dos seguintes critérios: taxa de filtração glomerular basal 30-59 ml/min, ECOG PS 2, perda de audição Grau 2 ou insuficiência cardíaca classe III da NYHA.
Os pacientes foram tratados na coorte de escalonamento de dose (n=5) e na coorte A (n=40) com pembrolizumabe 200 mg por 30 minutos no dia 1 e Padcev® 1,25 mg/kg nos dias 1 e 8 de cada ciclo de 21 dias, ou na coorte K (n = 76) onde pacientes foram estratificados pela ausência ou presença de metástases hepáticas e ECOG performance status e, em seguida, randomizados (1:1) para pembrolizumabe 200 mg por 30 minutos no dia 1 e Padcev® 1,25 mg/kg nos dias 1 e 8 de cada ciclo de 21 dias ou P Padcev® 1,25 mg/kg nos dias 1 e 8 de cada ciclo de 21 dias em monoterapia (n= 73).
O tratamento com Padcev® e pembrolizumabe continuou até a progressão da doença definida no RECIST vl.1, toxicidade inaceitável ou para pembrolizumabe um máximo de 35 ciclos de tratamento (aproximadamente 2 anos). O tratamento foi permitido além da progressão da doença definida no RECIST vl.1, se o investigador do tratamento considerasse que o paciente estivesse obtendo benefício clínico e o tratamento fosse tolerado. A avaliação do status do tumor foi realizada a cada 9 semanas por um ano e posteriormente a cada 12 semanas. As medidas de resultado de eficácia principais foram TRO e DR conforme avaliado pelo BICR utilizando RECIST vl.1.
As características da população do estudo eram: idade mediana de 71 anos, 74% homens e 85% brancos. Noventa e oito por cento tinham doença Ml e 3% tinham doença MO. Sessenta e três por cento tinham um tumor primário no trato inferior e 37% dos pacientes tinham um tumor primário no trato superior. Oitenta e quatro por cento dos pacientes tinham metástase visceral, incluindo 22% com metástase hepática. Entre os 121 pacientes, 108 pacientes foram avaliados para expressão de PPC, dos quais 57% eram PPC < 10 e 43% eram PPC 10. Sessenta por cento dos pacientes tinham depuração de creatinina basal < 60 ml/min e 30 ml/min, 85% tinham ECOG PS de 0-1 e 15% ECOG PS de 2. Trinta e nove por cento dos pacientes tinham histologia do carcinoma de células transicionais (CCT), 13% tinham CCT com diferenciação escamosa e 48% tinham CCT com outras variantes histológicas.
A mediana do tempo de acompanhamento para os pacientes tratados com Padcev® em combinação com pembrolizumabe no coorte K foi de 14,8 meses (intervalo: 0,6 a 26,2 meses) e para a coorte de escalonamento de dose e coorte A foi 44,7 meses (intervalo: 0,7 a 52,4 meses).
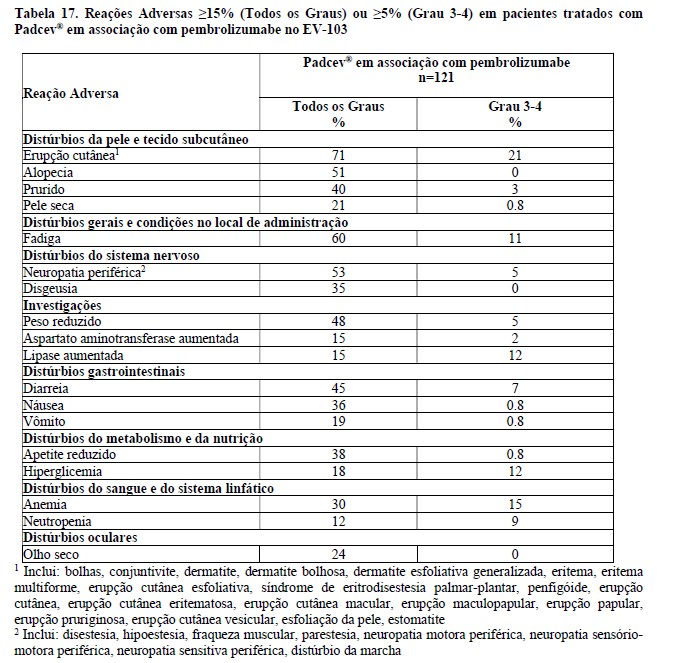
Os resultados de eficácia estão apresentados na tabela 4 abaixo. Na análise de eficácia combinada de escalonamento de dose/coorte A e coorte K (n = 121), TRO foi 68% (95% IC: 59, 76) com taxas de resposta completa e parcial de 12% e 55%, respectivamente. Entre os pacientes respondentes, 80 % tinham respostas de 6 meses ou mais (baseado na estimativa de Kaplan-Meier).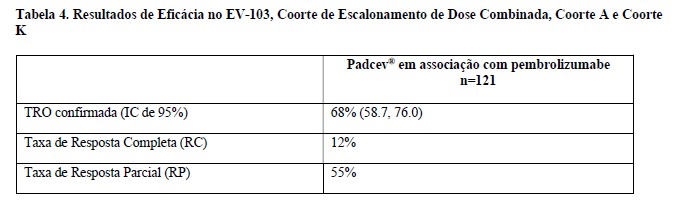
A duração mediana da resposta para a coorte de escalonamento de dose + Coorte A foi de 22,1 meses (intervalo: 1,0+ a 46,3+) e para a Coorte K não foi alcançada (intervalo: 1,2 a 24,1+).
Carcinoma Urotelial Localmente Avançado ou Metastático Anteriormente Não Tratado
Estudo Clínico EV-3024
A eficácia de Padcev® em combinação com pembrolizumabe foi avaliada no estudo EV-302, um ensaio multicêntrico aberto, randomizado, que envolveu 886 pacientes com câncer urotelial localmente avançado ou metastático que não receberam terapia sistêmica prévia para doença localmente avançada ou metastática. Pacientes com metástases ativas no SNC, neuropatia sensorial ou motora em curso de Grau ≥2, ou diabetes não controlada definida como hemoglobina A1C (HbA1c) ≥8% ou HbA1c ≥7% com sintomas de diabetes associados foram excluídos da participação no estudo.
Os pacientes foram randomizados 1:1 para receber Padcev® em combinação com pembrolizumabe ou quimioterapia à base de platina (gencitabina com cisplatina ou carboplatina). Os pacientes no braço Padcev® em combinação com pembrolizumabe receberam Padcev® 1,25 mg/kg como infusão intravenosa durante 30 minutos nos dias 1 e 8 de um ciclo de 21 dias, seguido de pembrolizumabe 200 mg no dia 1 aproximadamente 30 minutos após administração do Padcev® .
O tratamento foi continuado até a progressão da doença ou toxicidade inaceitável. Na ausência de progressão da doença ou toxicidade inaceitável, o pembrolizumabe foi continuado por até 2 anos. Os pacientes no braço de quimioterapia à base de platina receberam gencitabina 1.000 mg/m2 administrada nos dias 1 e 8 de um ciclo de 21 dias com cisplatina 70 mg/m2 ou carboplatina (AUC = 4,5 ou 5 de acordo com as diretrizes locais) administrada no dia 1 do ciclo de 21 dias. O tratamento foi continuado até a progressão da doença ou toxicidade inaceitável por até 6 ciclos. A randomização foi estratificada por elegibilidade para cisplatina, expressão de PD-L1 e presença de metástases hepáticas.
As características da população do estudo eram: idade mediana de 69 anos (intervalo: 22 a 91), 77% homens e 67% brancos, 22% eram asiáticos, 1% eram negros ou afro-americanos, e 10% eram desconhecidos ou de outra raça; 12% eram hispânicos ou latinos.
Os pacientes tinham um status de desempenho basal do ECOG de 0 (49%), 1 (47%) ou 2 (3%). Quarenta e sete por cento dos pacientes tinham uma HbA1c basal < 5,7%. No início do estudo, 95% dos pacientes tinham câncer urotelial metastático e 5% dos pacientes tinham câncer urotelial localmente avançado. Setenta e dois por cento dos pacientes apresentavam metástases viscerais no início do estudo, incluindo 23% com metástases hepáticas. Oitenta e cinco por cento dos pacientes apresentavam histologia de carcinoma urotelial (UC), incluindo 6% com diferenciação escamosa mista de UC e 2% com UC mista de outras variantes histológicas. Quarenta e seis por cento dos pacientes eram inelegíveis para cisplatina e 54% eram elegíveis para cisplatina no momento da randomização.
No momento da análise primária, 33% dos pacientes do braço Padcev® em combinação com pembrolizumabe e nenhum paciente do braço de quimioterapia à base de platina permaneciam em tratamento. Trinta e dois por cento dos pacientes no braço Padcev® em combinação com pembrolizumabe receberam terapia subsequente; 25% dos pacientes receberam quimioterapia à base de platina como primeira terapia subsequente. Setenta e um por cento dos pacientes no braço de quimioterapia à base de platina receberam terapia subsequente; 59% dos pacientes receberam um inibidor de PD-1 ou PD-L1, incluindo 30% dos pacientes que receberam terapia de manutenção com avelumabe, como primeira terapia subsequente.
O estudo demonstrou melhora estatisticamente significativa na sobrevida global (OS), na sobrevida livre de progressão (PFS) e na taxa de resposta objetiva (ORR) para pacientes randomizados para Padcev® em combinação com pembrolizumabe comparado com quimioterapia à base de platina (PFS e ORR foram avaliados por BICR usando RECIST v1.1). Os resultados de eficácia para pacientes que receberam Padcev® em combinação com pembrolizumabe foram consistentes em subgrupos pré-especificados, incluindo idade, região geográfica, ECOG PS inicial, local primário do tumor, metástase hepática, expressão de PD-1/PD-L1, elegibilidade para cisplatina e local da doença metastática.
O tempo médio de acompanhamento para este estudo foi de 17,2 meses (intervalo: 0,1 a 37,2).
A Tabela 5 e as Figuras 3 e 4 resumem os resultados de eficácia do estudo EV-302.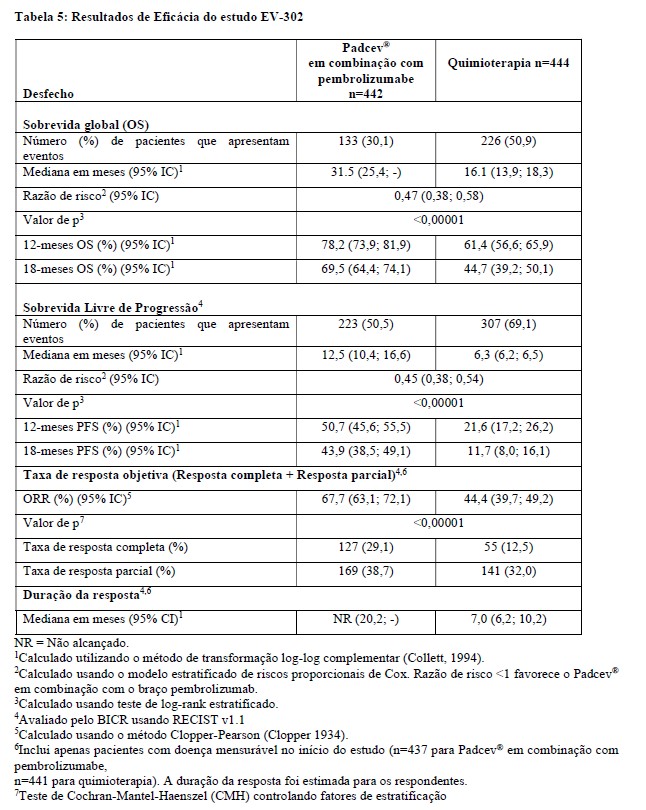
3. CARACTERÍSTICAS FARMACOLÓGICAS
Propriedades Farmacodinâmicas
Mecanismo de ação
O enfortumabe vedotina é um ADC. O anticorpo é um IgG1 kappa humano direcionado contra a nectina-4, uma proteína de adesão localizada na superfície das células. A molécula sintética, MMAE, é um agente de ruptura de microtúbulos, ligado ao anticorpo por meio de um ligante clivável por protease. Dados não clínicos sugerem que a atividade anticâncer de enfortumabe vedotina ocorre devido à ligação do ADC às células que expressam nectina-4, seguida pela internalização do complexo ADC-nectina-4 e a liberação do MMAE por meio de clivagem proteolítica. A liberação de MMAE interrompe a rede de microtúbulos dentro da célula, subsequentemente induzindo a parada do ciclo celular e apoptose e morte celular imunogênica. A combinação de enfortumabe vedotina com inibidores de PD-1 resulta em atividade antitumoral aumentada, consistente com os mecanismos complementares de citotoxicidade celular induzida por MMAE e indução de morte celular imunogênica, além da regulação positiva da função imune pela inibição de PD-1.
Efeitos farmacodinâmicos
Em uma análise de resposta à exposição para segurança, uma exposição mais elevada ao enfortumabe vedotina foi associada à maior incidência de algumas reações adversas (por exemplo, neuropatia periférica de grau ≥2, hiperglicemia de grau ≥3). A relação de exposição-resposta para a eficácia não foi totalmente caracterizada.
Eletrofisiologia Cardíaca
Na dose recomendada, Padcev® não causou grande prolongamento de intervalo QTc ( > 20 ms).
Propriedades Farmacocinéticas
A análise de farmacocinética da população incluiu os dados de 748 pacientes com base em estudos clinicos. A farmacocinética de enfortumabe vedotina como agente único foi caracterizada após doses únicas e múltiplas em pacientes com carcinoma urotelial localmente avançado ou metastático e outros tumores sólidos.
A farmacocinética do ADC e da MMAE não conjugada foi compatível quando avaliada após a administração de Padcev® como agente único e em associação com pembrolizumabe após 1 ciclo de tratamento.
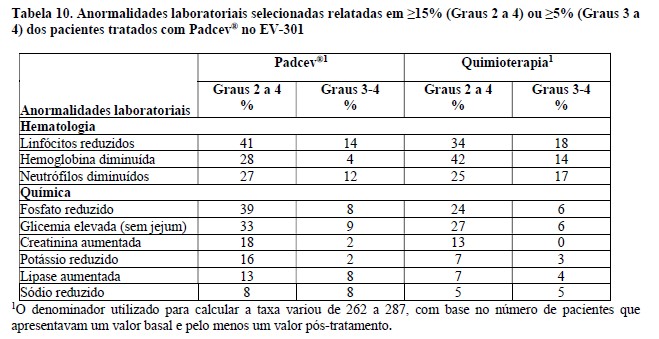
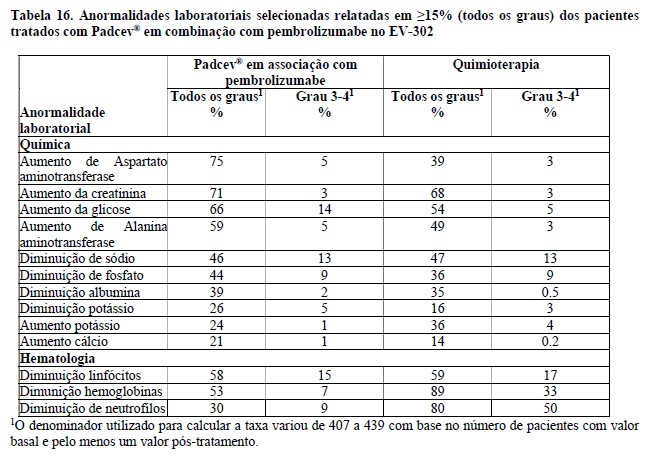
Os parâmetros de exposição do ADC e MMAE não conjugado (o componente citotóxico de enfortumabe vedotina) estão resumidos na Tabela 6 abaixo. As concentrações máximas do ADC foram observadas próximo do final da infusão intravenosa enquanto as concentrações máximas de MMAE não conjugada foram observadas aproximadamente 2 dias após a administração de enfortumabe vedotina. Um acúmulo mínimo de ADC e MMAE não conjugada foi observado após administrações repetidas de enfortumabe vedotina nos pacientes. As concentrações em estado de equilíbrio (steady-state) de ADC foram alcançadas após 1 ciclo de tratamento para enfortumabe vedotina como agente único e em combinação com pembrolizumabe. Para enfortumabe vedotina como agente único administrado em pacientes com carcinoma urotelial metastático ou localmente avançado previamente tratado, as concentrações de MMAE não conjugado pareceram atingir o estado de equilíbrio após 1 ciclo. Um declínio de 31% nas concentrações de MMAE não conjugado foi observado no final do ciclo 1 quando enfortumabe vedotina foi administrado como agente único e em combinação com pembrolizumabe em pacientes com doença localmente avançada ou mUC não tratados anteriormente.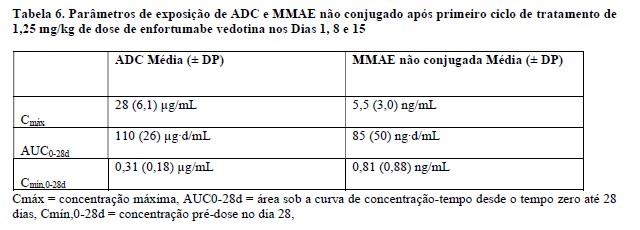
Distribuição
O volume de distribuição estimado em estado estacionário de ADC foi de 12,8 litros após a administração de enfortumabe vedotina. A ligação da proteína plasmática de MMAE não conjugado à proteína plasmática variou de 68 a82% in vitro.
Eliminação
ADC e MMAE não conjugado exibiram declínios multiexponenciais com meia-vida de eliminação de 3,6 dias e 2,6 dias, respectivamente. A depuração (CL=clearance) médio de enfortumabe vedotina e MMAE não conjugado em pacientes foi 0,11 L/h e 2,11 L/h, respectivamente. A eliminação de MMAE não conjugado pareceu ser limitada por sua taxa de liberação a partir de enfortumabe vedotina.
Metabolismo
O catabolismo de enfortumabe vedotina não foi estudado em humanos; no entanto, é esperado que sofra catabolismo para pequenos peptídeos, aminoácidos, MMAE não conjugado e catabólitos relacionados ao MMAE não conjugado. O enfortumabe vedotina libera MMAE não conjugado via clivagem proteolítica, e o MMAE não conjugado é metabolizado principalmente pela CYP3A4 in vitro.
Excreção
A excreção de enfortumabe vedotina não está totalmente caracterizada. Após uma dose única de outro ADC que contém MMAE não conjugado, 17% do MMAE não conjugado total administrado foram recuperados nas fezes e 6% na urina em um período de 1 semana, principalmente como droga inalterada. É esperado um perfil de excreção semelhante para MMAE não conjugado após a administração de enfortumabe vedotina.
Farmacocinética em populações especiais
Com base na análise farmacocinética populacional, não foram observadas diferenças clinicamente significativas na farmacocinética do enfortumabe vedotina com base na idade (24 a 90 anos), sexo ou raça/etnia (caucasianos, asiáticos, negros ou outros).
Insuficiência hepática
Com base na análise de farmacocinética populacional, houve um aumento de 37% e de 16% na exposição nas concentrações médias do MMAE não conjugado observado em pacientes previamente tratados e não tratados anteriormente com câncer localmente avançado ou mUC, respectivamente, com insuficiência hepática leve (bilirrubina total de 1 a 1,5 × LSN e AST qualquer, ou bilirrubina total ≤LSN e AST > LSN, n=65) em comparação a pacientes com função hepática normal. O enfortumabe vedotina foi estudado apenas em um número limitado de pacientes com insuficiência hepática moderada e não foi avaliado em pacientes com insuficiência hepática severa.
O efeito da insuficiência hepática moderada ou grave (bilirrubina total > 1,5 x LSN e AST qualquer) ou de transplante de fígado na farmacocinética de ADC ou MMAE não conjugado é desconhecido.
Insuficiência renal
A farmacocinética do enfortumabe vedotina e MMAE não conjugado foi avaliada após a administração de 1,25 mg/kg de enfortumabe vedotina a pacientes com insuficiência renal leve (depuração/clearance de creatinina: CrCL > 60-90 mL/min; n=272), moderada (CrCL 30-60 mL/min; n=315) e severa (CrCL < 30 mL/min; n=25). Não foram observadas diferenças significativas na exposição (AUC) de ADC ou MMAE não conjugada em pacientes com insuficiência renal leve, moderada ou grave em comparação a pacientes com função renal normal. O efeito da doença renal em estágio terminal com ou sem necessidade de diálise na farmacocinética do ADC ou MMAE não conjugado é desconhecido.
Uso geriátrico
A análise farmacocinética da população indica que a idade [intervalo: 24 a 90 anos; 60% (450/748) > 65 anos, 19% (143/748) > 75 anos] não tem efeito clinicamente significativo na farmacocinética de enfortumabe vedotina.
Populações especiais
Idosos
Não é necessário ajuste de dose em pacientes com idade superior a 65 anos. Vide item "Características farmacológicas".
Uso pediátrico
A segurança e a eficácia de enfortumabe vedotina não foram estabelecidas em pacientes pediátricos.
Pacientes com insuficiência renal
Não é exigido ajuste de dose em pacientes que apresentam insuficiência renal leve [depuração/clearance de creatinine (CrCL) > 60-90 mL/min], moderada (CrCL 30-60 mL/min) ou grave (CrCL < 30 mL/min). Enfortumabe vedotina não foi avaliado em pacientes com doença renal terminal. (vide item "Características farmacológicas").
Pacientes com insuficiência hepática
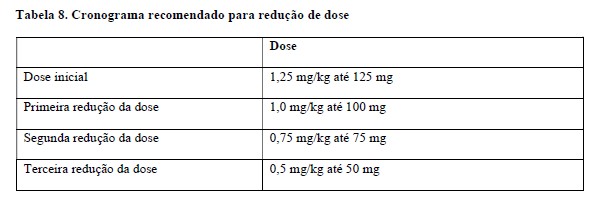
Evite o uso de Padcev® em pacientes com insuficiência hepática moderada ou severa (bilirrubina total > 1,5 x LSN e qualquer AST). Padcev® foi estudado apenas em um número limitado de pacientes com insuficiência hepática moderada (n=3) e não foi avaliado em pacientes com insuficiência hepática severa. Em outro ADC que contém MMAE, a frequência de reações adversas de grau ≥3 e óbitos foi maior em pacientes com insuficiência hepática moderada (Child-Pugh B) ou severa (Child-Pugh C) em comparação com pacientes com função hepática normal. Não é necessário ajuste da dose inicial ao administrar Padcev® a pacientes com insuficiência hepática leve (bilirrubina total 1 a 1,5 × LSN e qualquer AST, ou bilirrubina total ≤LSN e AST > LSN) (vide item "Características farmacológicas").
Dados de segurança não-clínica
Carcinogênese, mutagênese, comprometimento da fertilidade
Não foram realizados estudos de carcinogenicidade com enfortumabe vedotina ou com o agente citotóxico sintético (MMAE).
O MMAE foi genotóxico no estudo do micronúcleo da medula óssea de ratos por meio de um mecanismo aneugênico. Este efeito é compatível com o efeito farmacológico do MMAE como agente de ruptura dos microtúbulos. O MMAE não foi mutagênico no ensaio de mutação reversa bacteriana (teste de Ames) ou no ensaio de mutação direta de linfoma de camundongo L5178Y.
Não foram realizados estudos de fertilidade com enfortumabe vedotina ou MMAE. No entanto, os resultados dos estudos de toxicidade de doses repetidas realizados em ratos indicam o potencial de enfortumabe vedotina em comprometer a função reprodutiva e a fertilidade masculinas.
Em estudos de toxicologia de dose repetida realizados em ratos por até 13 semanas, as doses ≥2 mg/kg de enfortumabe vedotina (em exposições semelhantes às exposições na dose humana recomendada) resultaram em diminuições nos pesos dos testículos e do epidídimo, degeneração do túbulo seminífero, depleção de espermátides/espermatócitos nos testículos e detritos celulares, granuloma de esperma e hipospermia/espermátides anormais no epidídimo. Os achados nos testículos e epidídimo não foram revertidos até o final do período de recuperação.
Embora não observados com enfortumabe vedotina, efeitos ovarianos foram observados em estudos de toxicidade de dose repetida de outros ADCs contendo MMAE. Uma diminuição leve a moderada ou ausência de folículos ovarianos secundários e terciários foi observada em macacas cynomolgus fêmeas jovens em doses ≥3 mg/kg semanalmente por 4 semanas. Não foram observadas alterações nos folículos primordiais. Os efeitos nos folículos ovarianos secundários e terciários mostraram evidências de recuperação 6 semanas após o término da dosagem.
Não foram conduzidos estudos dedicados de segurança pré-clínica com enfortumabe vedotina em associação com pembrolizumabe.
4. CONTRAINDICAÇÕES
Padcev® é contraindicado a pacientes com hipersensibilidade a enfortumabe vedotina ou a quaisquer dos excipientes presentes na sua formulação.
Uso contraindicado no aleitamento ou na doação de leite humano.
Este medicamento é contraindicado durante o aleitamento ou doação de leite, pois pode ser excretado no leite humano e pode causar reações indesejáveis no bebê. Seu médico ou cirurgião-dentista deve apresentar alternativas para o seu tratamento ou para a alimentação do bebê.
5. ADVERTÊNCIAS E PRECAUÇÕES
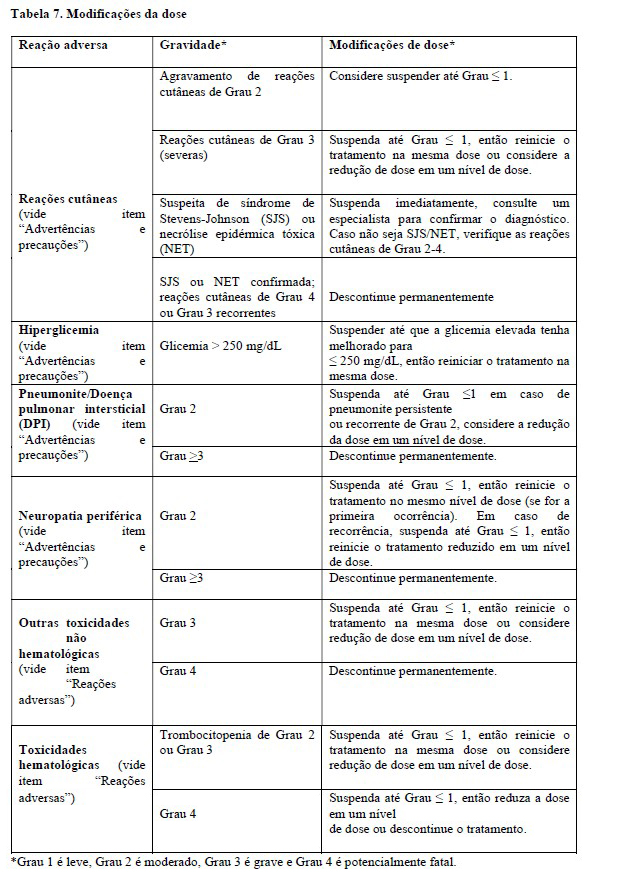
Reações cutâneas
Reações adversas cutâneas graves, incluindo casos fatais de síndrome de Stevens-Johnson (SSJ) ou necrólise epidérmica tóxica (NET), ocorreram em pacientes tratados com Padcev®. SSJ e NET ocorreram predominantemente durante o primeiro curso de tratamento, mas podem ocorrer mais tarde.
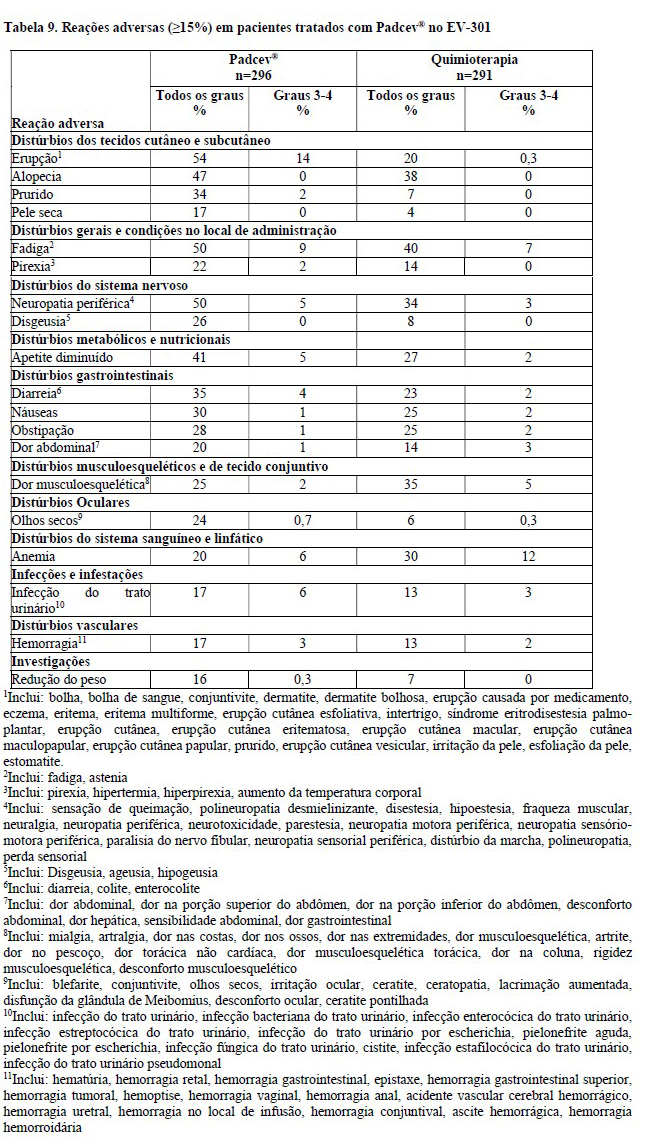
Reações cutâneas ocorreram em 57% dos 793 pacientes tratados com Padcev® como agente único em estudos clínicos. Vinte e quatro por cento (24%) dos pacientes apresentaram erupção maculopapulosa e 33% apresentaram coceira. Reações cutâneas de grau 3 a 4 ocorreram em 14% dos pacientes, incluindo erupção cutânea maculopapular, erupção cutânea eritematosa, erupção cutânea ou erupção medicamentosa, erupção intertriginosa e flexural simétrica relacionada ao fármaco (SDRIFE), dermatite bolhosa, dermatite esfoliativa, e eritrodisestesia palmoplantar. O tempo médio até o início das reações cutâneas graves foi de 0,7 meses (variação de 0,1 a 8 meses) Entre os pacientes que apresentaram reação cutânea que levou à interrupção da dose e que posteriormente retomaram a administração de Padcev® (n=80); 22% dos pacientes que reiniciaram com a mesma dose e 24% dos pacientes que reiniciaram com uma dose reduzida apresentaram reações cutâneas graves recorrentes. As reações cutâneas levaram à descontinuação do Padcev® em 3,2% dos pacientes (ver item "Reações adversas"). Dos pacientes que apresentaram reação cutânea e tiveram dados de resolução (N = 366), 61% tiveram resolução completa, 24% tiveram melhora parcial e 15% não tiveram melhora no momento da última avaliação. Dos 39% dos pacientes com reações cutâneas residuais na última avaliação, 38% apresentaram reações cutâneas de Grau ≥2.
Quando Padcev® foi administrado em associação com pembrolizumabe, a incidência de reações cutâneas, incluindo eventos graves, ocorreu em uma taxa mais alta em comparação com Padcev® como um único agente. Reações cutâneas ocorreram em 70% (todos os graus) dos 564 pacientes tratados com Padcev® em combinação com pembrolizumabe em estudos clínicos. A maioria das reações cutâneas que ocorreram com a terapia associada incluíram erupção cutânea maculopapular, erupção cutânea macular e erupção cutânea papular. Reações cutâneas de Grau 3-4 ocorreram em 17% dos pacientes (Grau 3: 16%, Grau 4: 1%), incluindo erupção cutânea maculopapular, dermatite bolhosa, dermatite, dermatite esfoliativa, penfigóide, erupção cutânea, erupção cutânea eritematosa, erupção cutânea macular e erupção papular. Uma reação fatal de dermatite bolhosa ocorreu em um paciente (0,2%). O tempo médio até o início das reações cutâneas graves foi de 1,7 meses e variou de 0,1 a 17,2 meses. As reações cutâneas levaram à descontinuação do Padcev® em 6% dos pacientes (ver item "Reações Adversas")".
Dos pacientes que apresentaram reação cutânea e tiveram dados de resolução (N = 391), 59% tiveram resolução completa, 30% tiveram melhora parcial e 10% não tiveram melhora no momento da última avaliação. Dos 41% dos pacientes com reações cutâneas residuais na última avaliação, 27% apresentaram reações cutâneas de Grau ≥2.
Monitore os pacientes de perto durante o tratamento de reações cutâneas. Considere o uso de corticosteroides tópicos e anti-histamínicos conforme indicação clínica.
Para reações cutâneas com piora de Grau 2, considere suspender Padcev® até Grau ≤1. Interrompa o Padcev® e encaminhe o paciente para tratamento com especialista se houver suspeita de SSJ, NET ou reações cutâneas graves (Grau 3). Descontinue Padcev® permanentemente em pacientes com SSJ ou NET confirmada; ou com reações cutâneas recorrentes de Grau 4 ou 3 (ver item "Posologia e modo de usar").
Hiperglicemia
Hiperglicemia e cetoacidose diabética (CAD), incluindo eventos fatais, ocorreram em pacientes com e sem diabetes mellitus preexistente tratados com Padcev®.
Os pacientes com hemoglobina AlC basal ≥8% foram excluídos dos estudos clínicos.
Em estudos clínicos de Padcev® como agente único, 17% dos 793 pacientes tratados com Padcev® desenvolveram hiperglicemia de qualquer grau; 7% dos pacientes desenvolveram hiperglicemia de grau 3 a 4 (Grau 3: 6.6%, Grau 4: 0.8%). Eventos fatais de hiperglicemia e cetoacidose diabética ocorreram em um paciente cada (0,1%). A incidência de hiperglicemia de grau 3 a 4 aumentou consistentemente em pacientes com índice de massa corporal mais alto e em pacientes com AlC basal mais alta. Cinco por cento (5%) dos pacientes necessitaram de início de terapia com insulina para tratar a hiperglicemia. O tempo médio até o início da hiperglicemia foi de 0,5 meses (range: 0 a 20 meses). A hiperglicemia levou à suspensão de Padcev® em 0,6% dos pacientes (ver item "Reações Adversas")".
Dos pacientes que apresentaram hiperglicemia e tiveram dados de resolução (N = 106), 66% tiveram resolução completa, 19% tiveram melhora parcial e 15% não tiveram melhora no momento da última avaliação. Dos 34% dos pacientes com hiperglicemia residual na última avaliação, 64% apresentavam hiperglicemia de Grau ≥2.
Monitore cuidadosamente os níveis de glicose no sangue em pacientes com, ou em risco de, diabetes mellitus ou hiperglicemia. Se a glicose no sangue estiver alta ( > 250 mg/dL), interrompa Padcev® (ver item "Posologia e modo de usar").
Pneumonite/Doença pulmonar intersticial (DPI)
Pneumonite/ Doença intersticial pulmonar (DPI), Pneumonite/DPI fatal grave ocorreu em pacientes tratados com Padcev®. Em estudos clínicos de Padcev® como agente único, 3% de 793 pacientes tratados com Padcev® apresentaram pneumonite/DIP de qualquer grau e 0,8% tiveram graus 3 a 4. O tempo até o início da pneumonite foi de 2,7 meses e variou de 0,6 a 6 meses.
A incidência de pneumonite/DIP, incluindo acontecimentos graves, ocorreu numa taxa mais elevada quando Padcev® foi administrado em combinação com pembrolizumabe em comparação com Padcev® como agente único. Quando Padcev® foi administrado em associação com pembrolizumabe, 10% dos 564 pacientes do Estudo EV-302 tratados com terapia de associação apresentaram pneumonite/DIP de qualquer grau e 4% apresentaram um evento grave de pneumonite. Pneumonite/DIP fatal ocorreu em dois pacientes (0,4%). O tempo mediano para o início da pneumonite/DIP foi de 4 meses, variando de 0,3 a 26 meses.
Monitore os pacientes quanto a sinais e sintomas indicativos de pneumonite/DIP, como hipóxia, tosse, dispneia ou infiltrados intersticiais em exames radiológicos. Avalie e exclua causas infecciosas, neoplásicas e outras para esses sinais e sintomas por meio de investigações apropriadas. Descontinue Padcev® em pacientes que desenvolverem pneumonite/DIP persistente ou recorrente de grau 2 e considere a redução da dose. Descontinue Padcev® permanentemente em todos os pacientes com pneumonite DII de grau 3 ou 4 (ver item "Posologia e modo de usar").
Neuropatia periférica
Neuropatia periférica ocorreu em 53% dos 793 pacientes tratados com Padcev® como agente único em estudos clínicos, incluindo 38% com neuropatia sensorial, 7% com fraqueza muscular e 7% com neuropatia motora, trinta porcento dos pacientes apresentaram reações de Grau 2 e 5% tiveram reações de grau 3 a 4. Neuropatia periférica ocorreu em pacientes tratados com Padcev® com ou sem neuropatia periférica preexistente. O tempo até o início da neuropatia periférica de Grau ≥2 variou de 0,1 a 20 meses (mediana de 4,9 meses). A neuropatia levou à descontinuação do tratamento em 7% dos pacientes (ver reações Adversas). Dos pacientes que apresentaram neuropatia que tiveram dados sobre resolução (N = 340), 14% tiveram resolução completa, 46% tiveram melhora parcial e 41% não tiveram melhora no momento de sua última avaliação. Dos 86% dos pacientes com neuropatia residual na última avaliação, 51% apresentavam neuropatia Grau ≥ 2 ou maior.
A incidência de neuropatia periférica ocorreu em uma taxa mais alta quando Padcev® foi administrado em combinação com pembrolizumabe comparado com Padcev® como agente único. Quando Padcev® foi administrado em combinação com pembrolizumabe, 67% dos 564 pacientes tratados com terapia combinada apresentaram neuropatia periférica de qualquer grau, 36% neuropatia de grau 2 e 7% neuropatia de grau 3. O tempo médio para o início da neuropatia periférica de Grau ≥:2 foi de 6 meses (intervalo: 0,3 a 25 meses) (ver Reações Adversas).
Dos pacientes que apresentaram neuropatia e tiveram dados de resolução (N = 373), 13% tiveram resolução completa, 42% tiveram melhora parcial e 46% não tiveram melhora no momento da última avaliação. Dos 87% dos pacientes com neuropatia residual na última avaliação, 45% apresentavam neuropatia de Grau ≥2.
Monitore os pacientes quanto ao aparecimento ou piora dos sintomas de neuropatia periférica e considere a interrupção ou redução da dose de Padcev® em caso de neuropatia periférica. Descontinue Padcev® permanentemente em todos os pacientes que desenvolverem neuropatia periférica de Grau ≥3 (consultar item "Posologia e modo de usar").
Distúrbios oculares
Os distúrbios oculares foram relatados em 40% dos 384 pacientes tratados com Padcev® como agente único em estudos clínicos nos quais exames oftalmológicos estavam programados. A maioria destes eventos envolveu a córnea e incluiu eventos associados com olhos secos, como ceratite, visão turva, aumento da lacrimação, conjuntivite, deficiência de células-tronco límbicas e ceratopatia.
Os sintomas de olhos secos ocorreram em 30% dos pacientes, e visão turva ocorreu em 10% dos pacientes durante o tratamento com Padcev®. O tempo mediano até o início do distúrbio ocular sintomático foi 2,1 meses (variação: 0 a 22,4 meses). Monitore os pacientes quanto a distúrbios oculares. Considere o uso de colírio como medida profilática para profilaxia de olho seco e a realização de avaliação oftalmológica se os sintomas oculares ocorrerem ou não se resolverem. Considere o tratamento com esteroides oftálmicos tópicos, se indicado após um exame oftalmológico. Considere a interrupção da dose ou redução da dose de Padcev® em caso de distúrbios oculares sintomáticos.
Extravasamento no local da infusão
Reações de pele e tecidos moles secundárias ao extravasamento foram observadas após a administração de Padcev®. Dos 793 pacientes tratados com Padcev® como agente único em ensaios clínicos, 1,4% apresentaram reações cutâneas e de tecidos moles, incluindo 0,3% que apresentaram reações de grau 3 a 4. As reações podem ocorrer tardiamente. Eritema, edema, eleva